

The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote
- PMID: 15175259
- PMCID: PMC420352
- DOI: 10.1101/gad.1198204
The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote
Abstract
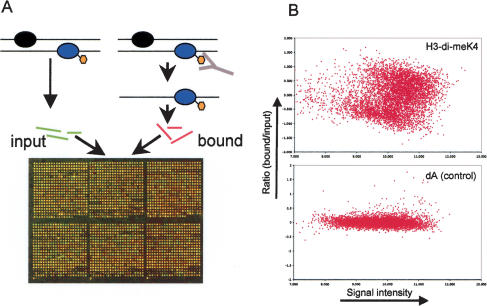
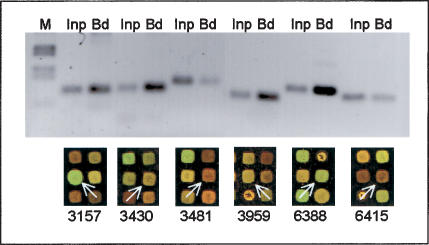
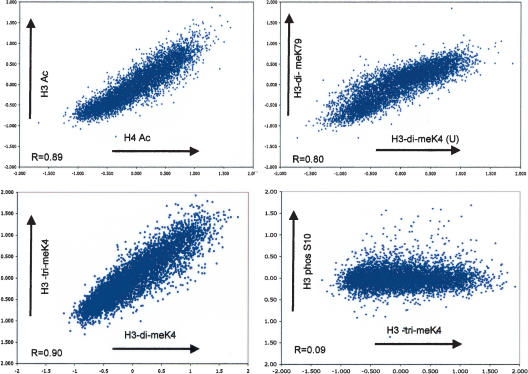
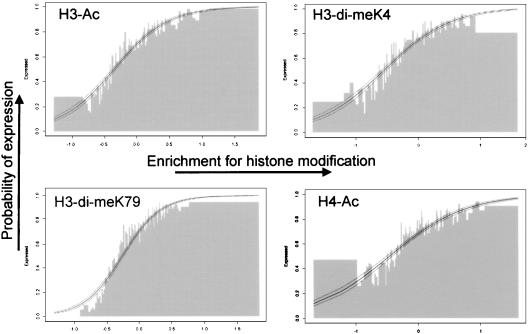
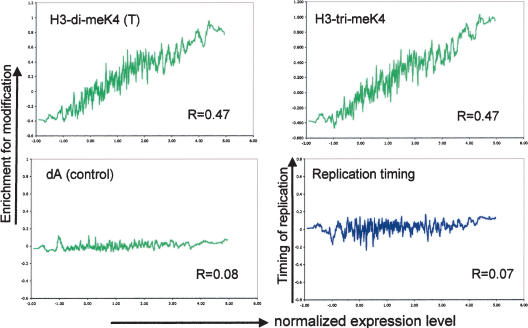
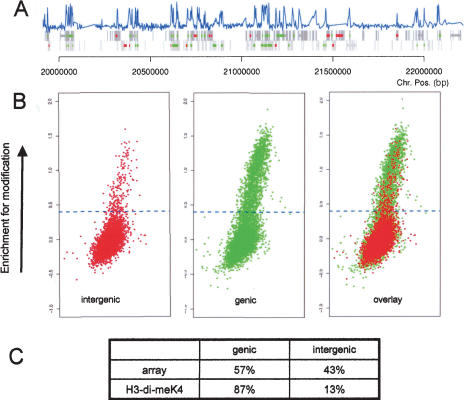
The covalent modification of nucleosomal histones has emerged as a major determinant of chromatin structure and gene activity. To understand the interplay between various histone modifications, including acetylation and methylation, we performed a genome-wide chromatin structure analysis in a higher eukaryote. We found a binary pattern of histone modifications among euchromatic genes, with active genes being hyperacetylated for H3 and H4 and hypermethylated at Lys 4 and Lys 79 of H3, and inactive genes being hypomethylated and deacetylated at the same residues. Furthermore, the degree of modification correlates with the level of transcription, and modifications are largely restricted to transcribed regions, suggesting that their regulation is tightly linked to polymerase activity.
Figures
References
-
- Adams, M.D., Celniker, S.E., Holt, R.A., Evans, C.A., Gocayne, J.D., Amanatides, P.G., Scherer, S.E., Li, P.W., Hoskins, R.A., Galle, R.F., et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185-2195. - PubMed
-
- Ahmad, K. and Henikoff, S. 2002a. Epigenetic consequences of nucleosome dynamics. Cell 111: 281-284. - PubMed
-
- ____. 2002b. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9: 1191-1200. - PubMed
-
- Bannister, A.J., Schneider, R., and Kouzarides, T. 2002. Histone methylation: Dynamic or static? Cell 109: 801-806. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases