

p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress
- PMID: 15181148
- PMCID: PMC515331
- DOI: 10.1091/mbc.e03-12-0871
p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress
Abstract
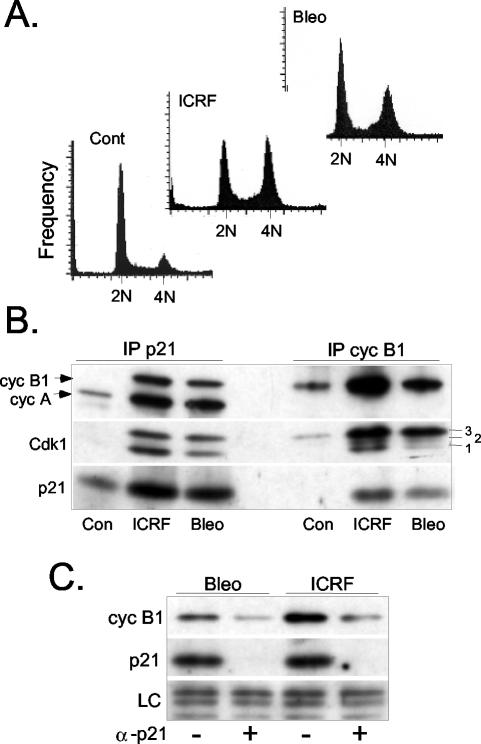
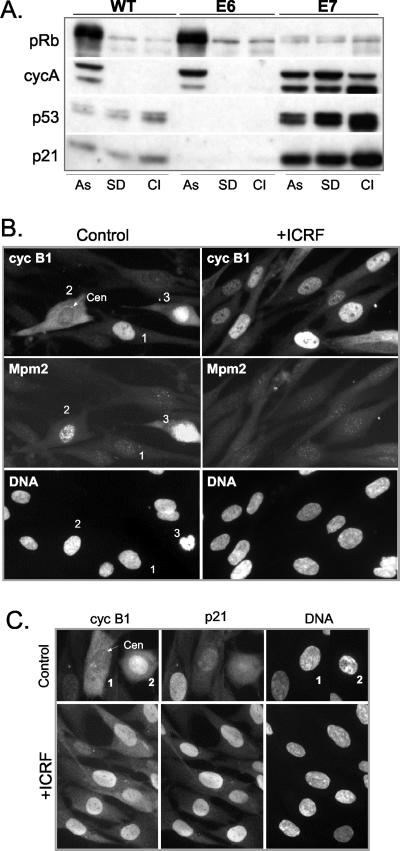
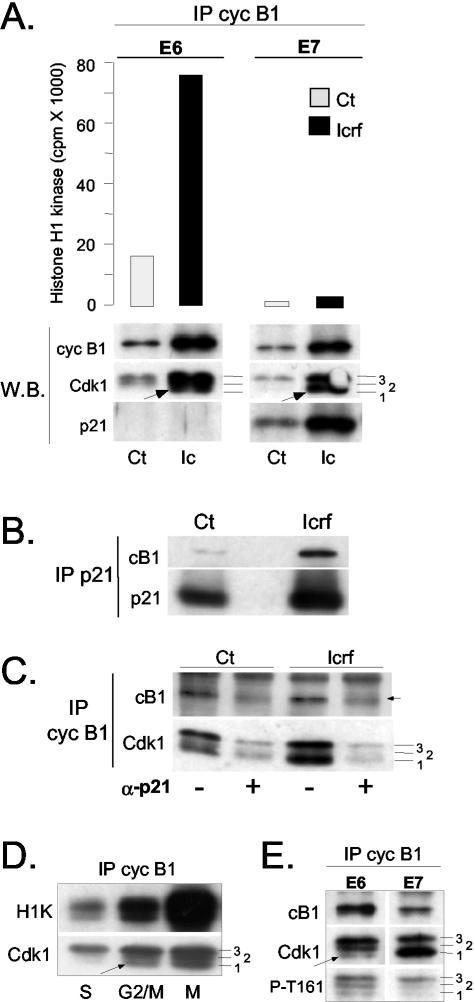
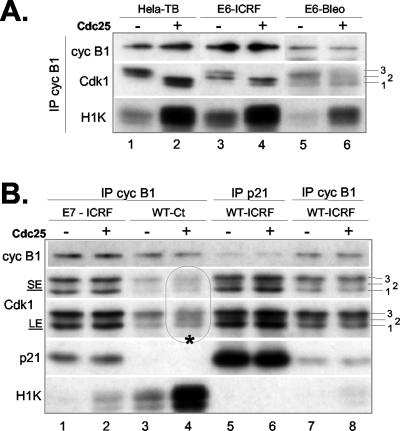
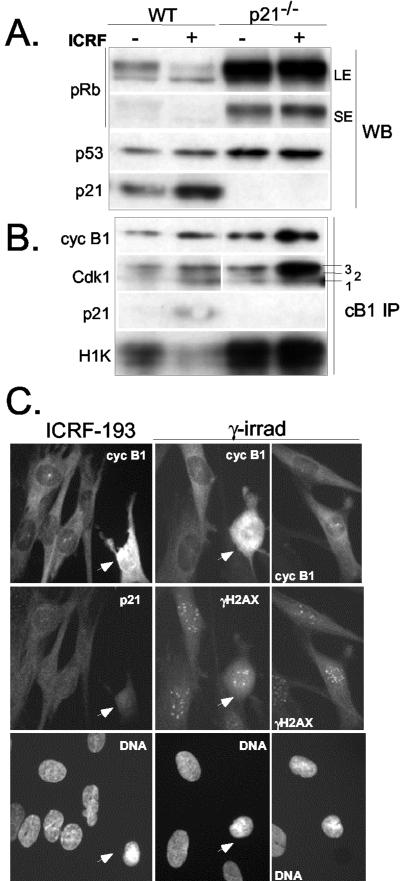
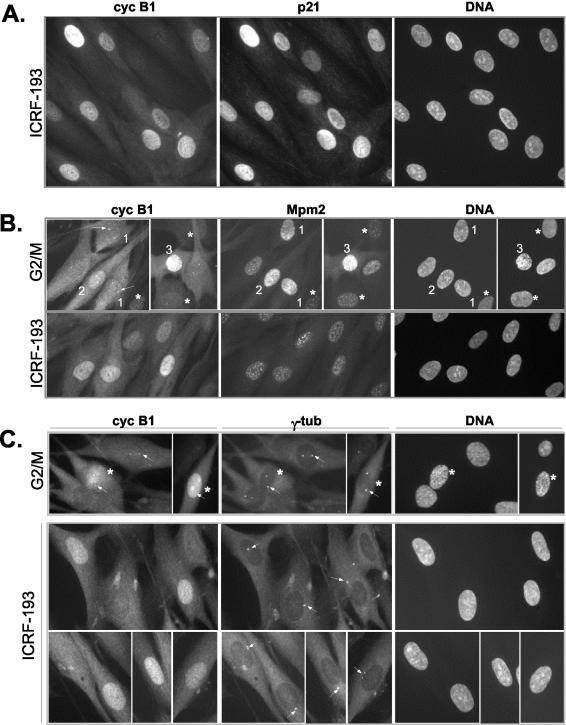
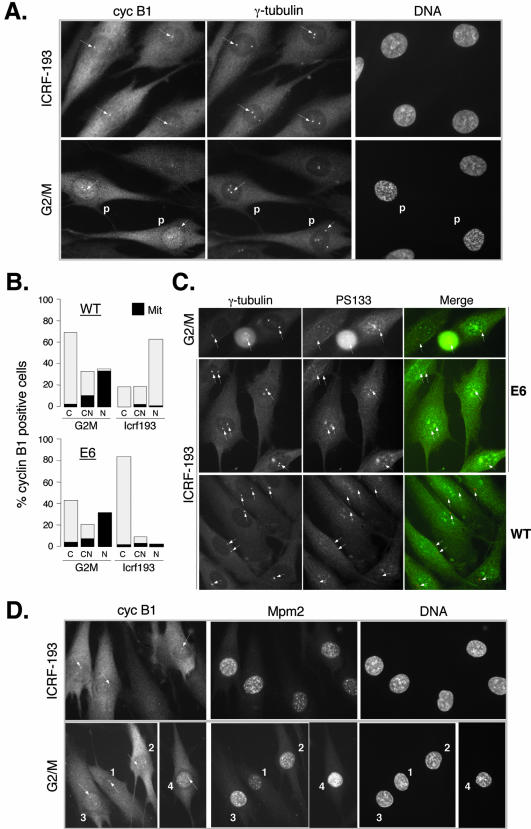
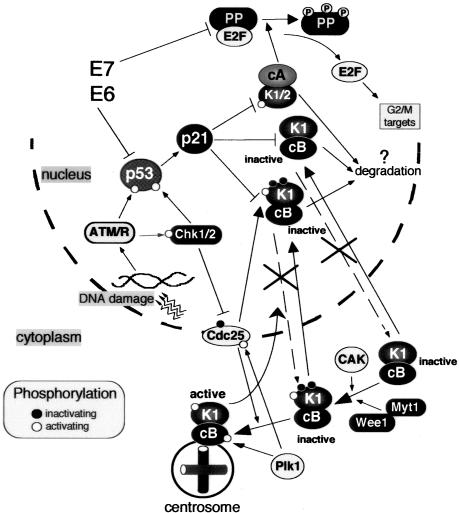
G2 arrest of cells suffering DNA damage in S phase is crucial to avoid their entry into mitosis, with the concomitant risks of oncogenic transformation. According to the current model, signals elicited by DNA damage prevent mitosis by inhibiting both activation and nuclear import of cyclin B1-Cdk1, a master mitotic regulator. We now show that normal human fibroblasts use additional mechanisms to block activation of cyclin B1-Cdk1. In these cells, exposure to nonrepairable DNA damage leads to nuclear accumulation of inactive cyclin B1-Cdk1 complexes. This nuclear retention, which strictly depends on association with endogenous p21, prevents activation of cyclin B1-Cdk1 by Cdc25 and Cdk-activating kinase as well as its recruitment to the centrosome. In p21-deficient normal human fibroblasts and immortal cell lines, cyclin B1 fails to accumulate in the nucleus and could be readily detected at the centrosome in response to DNA damage. Therefore, in normal cells, p21 exerts a dual role in mediating DNA damage-induced cell cycle arrest and exit before mitosis. In addition to blocking pRb phosphorylation, p21 directly prevents mitosis by inactivating and maintaining the inactive state of mitotic cyclin-Cdk complexes. This, with subsequent degradation of mitotic cyclins, further contributes to the establishment of a permanent G2 arrest.
Figures
References
-
- Aprelikova, O., Xiong, Y., and Liu, E.T. (1995). Both p16 and p21 families of cyclin-dependent kinase (CDK) inhibitors block the phosphorylation of cyclin-dependent kinases by the CDK-activating kinase. J. Biol. Chem. 270, 18195-18197. - PubMed
-
- Barboule, N., Lafon, C., Chadebech, P., Vidal, S., and Valette, A. (1999). Involvement of p21 in the PKC-induced regulation of the G2/M cell cycle transition. FEBS Lett. 444, 32-37. - PubMed
-
- Berry, L.D., and Gould, K.L. (1996). Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog. Cell Cycle Res. 2, 99-105. - PubMed
-
- Borgne, A., and Meijer, L. (1996). Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 271, 27847-27854. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
