

A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases
- PMID: 15189570
- PMCID: PMC446185
- DOI: 10.1186/1471-2105-5-76
A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases
Abstract
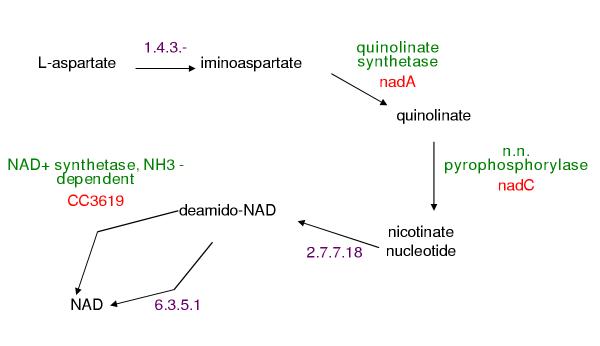
Background: The PathoLogic program constructs Pathway/Genome databases by using a genome's annotation to predict the set of metabolic pathways present in an organism. PathoLogic determines the set of reactions composing those pathways from the enzymes annotated in the organism's genome. Most annotation efforts fail to assign function to 40-60% of sequences. In addition, large numbers of sequences may have non-specific annotations (e.g., thiolase family protein). Pathway holes occur when a genome appears to lack the enzymes needed to catalyze reactions in a pathway. If a protein has not been assigned a specific function during the annotation process, any reaction catalyzed by that protein will appear as a missing enzyme or pathway hole in a Pathway/Genome database.
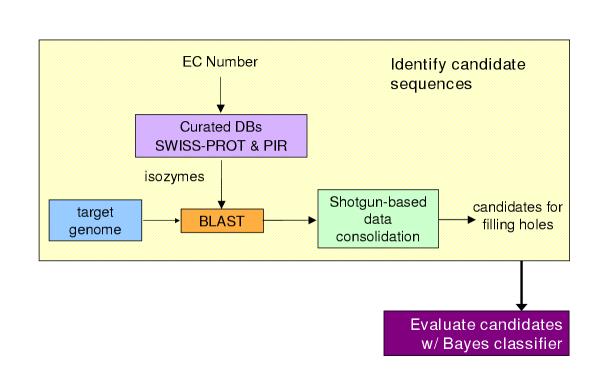
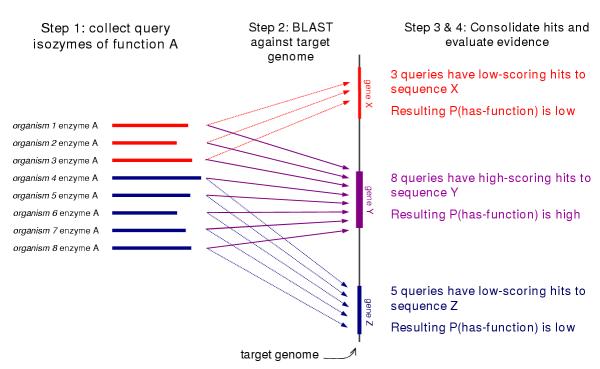
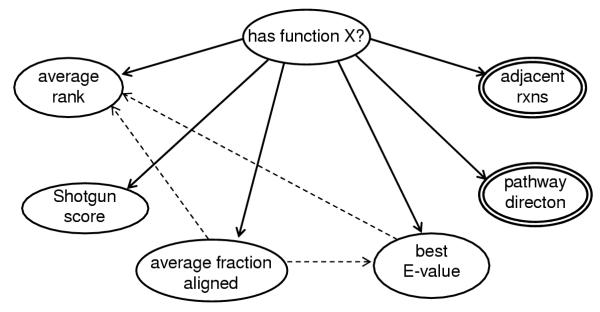
Results: We have developed a method that efficiently combines homology and pathway-based evidence to identify candidates for filling pathway holes in Pathway/Genome databases. Our program not only identifies potential candidate sequences for pathway holes, but combines data from multiple, heterogeneous sources to assess the likelihood that a candidate has the required function. Our algorithm emulates the manual sequence annotation process, considering not only evidence from homology searches, but also considering evidence from genomic context (i.e., is the gene part of an operon?) and functional context (e.g., are there functionally-related genes nearby in the genome?) to determine the posterior belief that a candidate has the required function. The method can be applied across an entire metabolic pathway network and is generally applicable to any pathway database. The program uses a set of sequences encoding the required activity in other genomes to identify candidate proteins in the genome of interest, and then evaluates each candidate by using a simple Bayes classifier to determine the probability that the candidate has the desired function. We achieved 71% precision at a probability threshold of 0.9 during cross-validation using known reactions in computationally-predicted pathway databases. After applying our method to 513 pathway holes in 333 pathways from three Pathway/Genome databases, we increased the number of complete pathways by 42%. We made putative assignments to 46% of the holes, including annotation of 17 sequences of previously unknown function.
Conclusions: Our pathway hole filler can be used not only to increase the utility of Pathway/Genome databases to both experimental and computational researchers, but also to improve predictions of protein function.
Figures
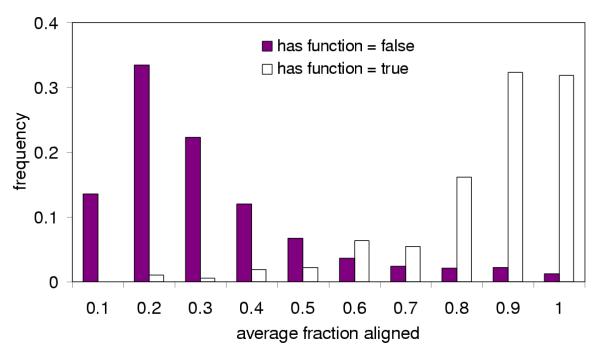
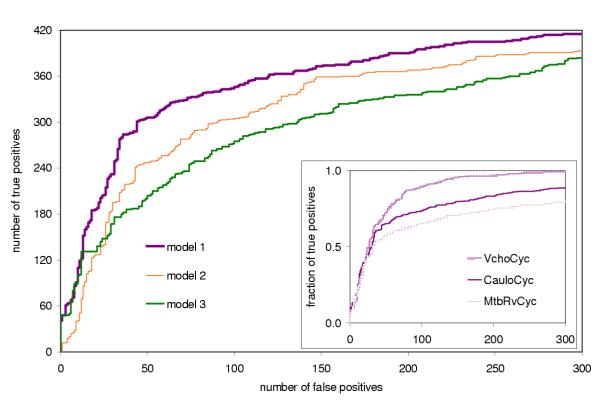
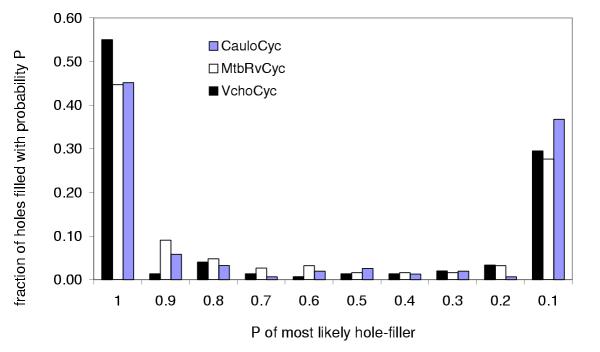
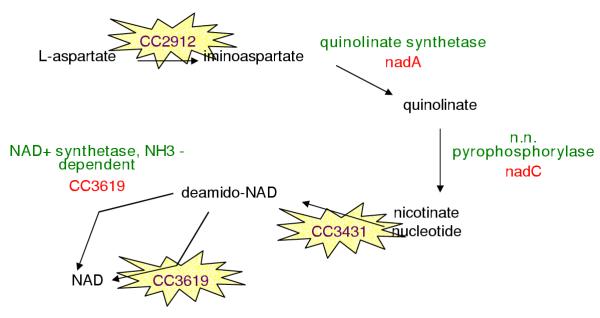
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
