

Sequence analysis of the genome of the Neodiprion sertifer nucleopolyhedrovirus
- PMID: 15194780
- PMCID: PMC421636
- DOI: 10.1128/JVI.78.13.7036-7051.2004
Sequence analysis of the genome of the Neodiprion sertifer nucleopolyhedrovirus
Abstract
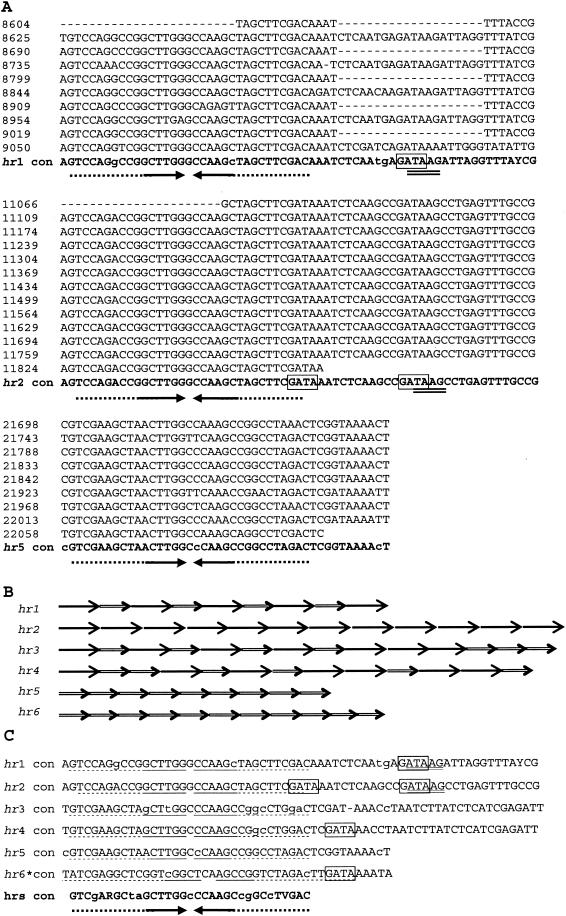
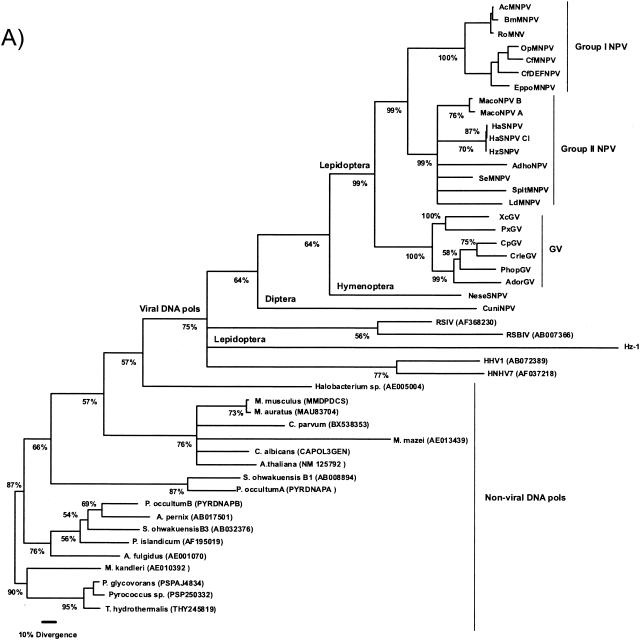
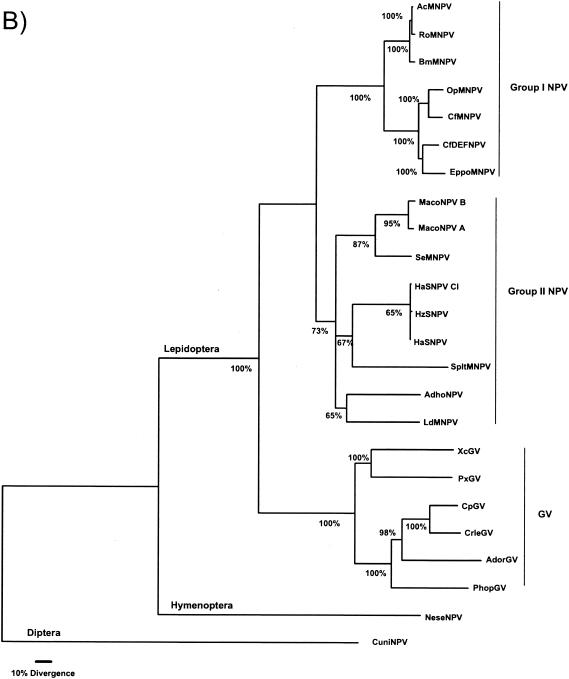
The genome of the Neodiprion sertifer nucleopolyhedrovirus (NeseNPV), which infects the European pine sawfly, N. sertifer (Hymenoptera: Diprionidae), was sequenced and analyzed. The genome was 86,462 bp in size. The C+G content of 34% was lower than that of the majority of baculoviruses. A total of 90 methionine-initiated open reading frames (ORFs) with more than 50 amino acids and minimal overlapping were found. From those, 43 ORFs were homologous to other baculovirus ORFs, and 29 of these were from the 30 conserved core genes among all baculoviruses. A NeseNPV homolog to the ld130 gene, which is present in all other baculovirus genomes sequenced to date, could not be identified. Six NeseNPV ORFs were similar to non-baculovirus-related genes, one of which was a trypsin-like gene. Only one iap gene, containing a single BIR motif and a RING finger, was found in NeseNPV. Two NeseNPV ORFs (nese18 and nese19) were duplicates transcribed in opposite orientations from each other. NeseNPV did not have an AcMNPV ORF 2 homolog characterized as the baculovirus repeat ORF (bro). Six homologous regions (hrs) were located within the NeseNPV genome, each containing small palindromes embedded within direct repeats. A phylogenetic analysis was done to root the tree based upon the sequences of DNA polymerase genes of NeseNPV, 23 other baculoviruses, and other phyla. Baculovirus phylogeny was then constructed with 29 conserved genes from 24 baculovirus genomes. Culex nigripalpus nucleopolyhedrovirus (CuniNPV) was the most distantly related baculovirus, branching to the hymenopteran NeseNPV and the lepidopteran nucleopolyhedroviruses and granuloviruses.
Figures


References
-
- Ahrens, C. H., R. L. Russell, C. J. Funk, J. T. Evans, S. H. Harwood, and G. F. Rohrmann. 1997. The sequence of the Orgyia pseudotsugata multinucleocapsid nuclear polyhedrosis virus genome. Virology 229:381-399. - PubMed
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. - PubMed
-
- Ayres, M. C., S. C. Howard, J. Kuzio, M. Lopez-Ferber, and R. D. Possee. 1994. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 202:586-605. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
