

RNAProfile: an algorithm for finding conserved secondary structure motifs in unaligned RNA sequences
- PMID: 15199174
- PMCID: PMC434454
- DOI: 10.1093/nar/gkh650
RNAProfile: an algorithm for finding conserved secondary structure motifs in unaligned RNA sequences
Abstract
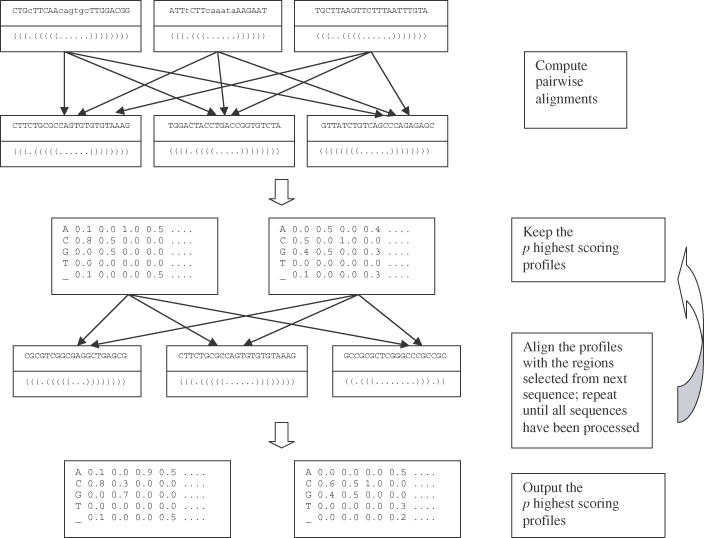
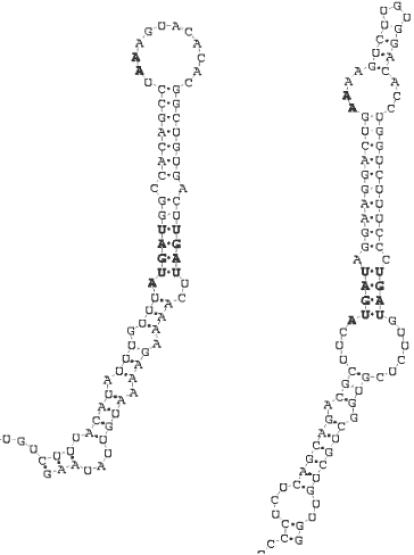
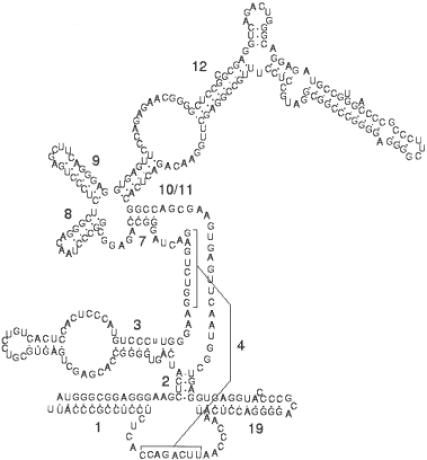
The recent interest sparked due to the discovery of a variety of functions for non-coding RNA molecules has highlighted the need for suitable tools for the analysis and the comparison of RNA sequences. Many trans-acting non-coding RNA genes and cis-acting RNA regulatory elements present motifs, conserved both in structure and sequence, that can be hardly detected by primary sequence analysis alone. We present an algorithm that takes as input a set of unaligned RNA sequences expected to share a common motif, and outputs the regions that are most conserved throughout the sequences, according to a similarity measure that takes into account both the sequence of the regions and the secondary structure they can form according to base-pairing and thermodynamic rules. Only a single parameter is needed as input, which denotes the number of distinct hairpins the motif has to contain. No further constraints on the size, number and position of the single elements comprising the motif are required. The algorithm can be split into two parts: first, it extracts from each input sequence a set of candidate regions whose predicted optimal secondary structure contains the number of hairpins given as input. Then, the regions selected are compared with each other to find the groups of most similar ones, formed by a region taken from each sequence. To avoid exhaustive enumeration of the search space and to reduce the execution time, a greedy heuristic is introduced for this task. We present different experiments, which show that the algorithm is capable of characterizing and discovering known regulatory motifs in mRNA like the iron responsive element (IRE) and selenocysteine insertion sequence (SECIS) stem-loop structures. We also show how it can be applied to corrupted datasets in which a motif does not appear in all the input sequences, as well as to the discovery of more complex motifs in the non-coding RNA.
Figures







Similar articles
-
De novo secondary structure motif discovery using RNAProfile.Methods Mol Biol. 2015;1269:49-62. doi: 10.1007/978-1-4939-2291-8_4. Methods Mol Biol. 2015. PMID: 25577372
-
A graph theoretical approach for predicting common RNA secondary structure motifs including pseudoknots in unaligned sequences.Bioinformatics. 2004 Jul 10;20(10):1591-602. doi: 10.1093/bioinformatics/bth131. Epub 2004 Feb 12. Bioinformatics. 2004. PMID: 14962926
-
MoD Tools: regulatory motif discovery in nucleotide sequences from co-regulated or homologous genes.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W566-70. doi: 10.1093/nar/gkl285. Nucleic Acids Res. 2006. PMID: 16845071 Free PMC article.
-
Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis.Biochimie. 2002 Aug;84(8):765-74. doi: 10.1016/s0300-9084(02)01405-0. Biochimie. 2002. PMID: 12457564 Review.
-
A systematic analysis of disease-associated variants in the 3' regulatory regions of human protein-coding genes II: the importance of mRNA secondary structure in assessing the functionality of 3' UTR variants.Hum Genet. 2006 Oct;120(3):301-33. doi: 10.1007/s00439-006-0218-x. Epub 2006 Jun 29. Hum Genet. 2006. PMID: 16807757 Review.
Cited by
-
RScan: fast searching structural similarities for structured RNAs in large databases.BMC Genomics. 2007 Jul 31;8:257. doi: 10.1186/1471-2164-8-257. BMC Genomics. 2007. PMID: 17663795 Free PMC article.
-
Can Clustal-style progressive pairwise alignment of multiple sequences be used in RNA secondary structure prediction?BMC Bioinformatics. 2007 Jun 8;8:190. doi: 10.1186/1471-2105-8-190. BMC Bioinformatics. 2007. PMID: 17559658 Free PMC article.
-
Identification of consensus RNA secondary structures using suffix arrays.BMC Bioinformatics. 2006 May 5;7:244. doi: 10.1186/1471-2105-7-244. BMC Bioinformatics. 2006. PMID: 16677380 Free PMC article.
-
Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers.Bioinformatics. 2012 Jun 15;28(12):i215-23. doi: 10.1093/bioinformatics/bts210. Bioinformatics. 2012. PMID: 22689764 Free PMC article.
-
RNA motif discovery: a computational overview.Biol Direct. 2015 Oct 9;10:61. doi: 10.1186/s13062-015-0090-5. Biol Direct. 2015. PMID: 26453353 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
