

Clinical and pathological characteristics of four Korean patients with limb-girdle muscular dystrophy type 2B
- PMID: 15201514
- PMCID: PMC2816849
- DOI: 10.3346/jkms.2004.19.3.447
Clinical and pathological characteristics of four Korean patients with limb-girdle muscular dystrophy type 2B
Abstract
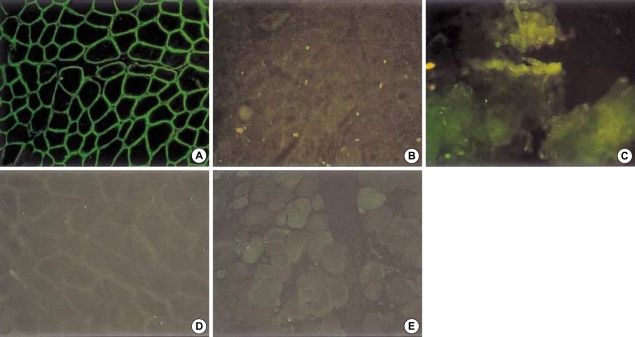
Limb-girdle muscular dystrophy type 2B (LGMD2B), a subtype of autosomal recessive limb-girdle muscular dystrophy (ARLGMD), is characterized by a relatively late onset and slow progressive course. LGMD2B is known to be caused by the loss of the dysferlin protein at sarcolemma in muscle fibers. In this study, the clinical and pathological characteristics of Korean LGMD2B patients were investigated. Seventeen patients with ARLGMD underwent muscle biopsy and the histochemical examination was performed. For the immunocytochemistry, a set of antibodies against dystrophin, alpha, beta, gamma, delta-sarcoglycans, dysferlin, caveolin-3, and beta-dystroglycan was used. Four patients (24%) showed selective loss of immunoreactivity against dysferlin at the sarcolemma on the muscle specimens. Therefore, they were classified into the LGMD2B category. The age at the onset of disease ranged from 9 yr to 33 yr, and none of the patients was wheelchair bound at the neurological examination. The serum creatine kinase (CK) was high in all the patients (4010-5310 IU/L). The pathologic examination showed mild to moderate dystrophic features. These are the first Korean LGMD2B cases with a dysferlin deficiency confirmed by immunocytochemistry. The clinical, pathological, and immunocytochemical findings of the patients with LGMD2B in this study were in accordance with those of other previous reports.
Figures


Similar articles
-
Dysferlin protein analysis in limb-girdle muscular dystrophies.J Mol Neurosci. 2001 Aug;17(1):71-80. doi: 10.1385/JMN:17:1:71. J Mol Neurosci. 2001. PMID: 11665864
-
[Dysferlin expression in limb-girdle muscular dystrophy and Miyoshi myopathy: analysis of 45 cases].Zhonghua Yi Xue Za Zhi. 2007 Jun 5;87(21):1486-90. Zhonghua Yi Xue Za Zhi. 2007. PMID: 17785089 Chinese.
-
Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G.Am J Med Genet. 1999 Feb 19;82(5):392-8. doi: 10.1002/(sici)1096-8628(19990219)82:5<392::aid-ajmg7>3.0.co;2-0. Am J Med Genet. 1999. PMID: 10069710
-
[Mutational and clinical features of Japanese patients with dysferlinopathy (Miyoshi myopathy and limb girdle muscular dystrophy type 2B)].Rinsho Shinkeigaku. 2005 Nov;45(11):938-42. Rinsho Shinkeigaku. 2005. PMID: 16447768 Review. Japanese.
-
Limb-Girdle Muscular Dystrophy 2B and Miyoshi Presentations of Dysferlinopathy.Am J Med Sci. 2017 May;353(5):484-491. doi: 10.1016/j.amjms.2016.05.024. Epub 2016 May 30. Am J Med Sci. 2017. PMID: 28502335 Review.
Cited by
-
Heterogeneous characteristics of Korean patients with dysferlinopathy.J Korean Med Sci. 2012 Apr;27(4):423-9. doi: 10.3346/jkms.2012.27.4.423. Epub 2012 Mar 21. J Korean Med Sci. 2012. PMID: 22468107 Free PMC article.
-
Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations.Eur J Hum Genet. 2011 Sep;19(9):974-80. doi: 10.1038/ejhg.2011.70. Epub 2011 Apr 27. Eur J Hum Genet. 2011. PMID: 21522182 Free PMC article.
-
Atypical Miyoshi distal myopathy: A case report.Exp Ther Med. 2016 Nov;12(5):3068-3072. doi: 10.3892/etm.2016.3716. Epub 2016 Sep 20. Exp Ther Med. 2016. PMID: 27882118 Free PMC article.
-
Anesthetic management of a patient with limb-girdle muscular dystrophy 2B:CARE-compliant case report and literature review.BMC Anesthesiol. 2019 Aug 17;19(1):155. doi: 10.1186/s12871-019-0813-8. BMC Anesthesiol. 2019. PMID: 31421689 Free PMC article. Review.
References
-
- Zatz M, de Paula F, Starling A, Vainzof M. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord. 2003;13:532–544. - PubMed
-
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH., Jr Dysferlin, a novel skeletal muscle genes, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. - PubMed
-
- Britton S, Freeman T, Vafiadaki E, Keers S, Harrison R, Bushby K, Bashir R. The third human FER-1-like protein is highly similar to dysferlin. Genomics. 2000;68:313–321. - PubMed
-
- Matsuda C, Aoki M, Hayashi YK, Ho MF, Arahata K, Brown RH., Jr Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology. 1999;53:1119–1122. - PubMed
-
- Anderson LV, Davison K, Moss JA, Young C, Cullen MJ, Walsh J, Johnson MA, Bashir R, Britton S, Keers S, Argov Z, Mahjneh I, Fougerousse F, Beckmann JS, Bushby KM. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum Mol Genet. 1999;8:855–861. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
