

Improved protein structure selection using decoy-dependent discriminatory functions
- PMID: 15207004
- PMCID: PMC449718
- DOI: 10.1186/1472-6807-4-8
Improved protein structure selection using decoy-dependent discriminatory functions
Abstract
Background: A key component in protein structure prediction is a scoring or discriminatory function that can distinguish near-native conformations from misfolded ones. Various types of scoring functions have been developed to accomplish this goal, but their performance is not adequate to solve the structure selection problem. In addition, there is poor correlation between the scores and the accuracy of the generated conformations.
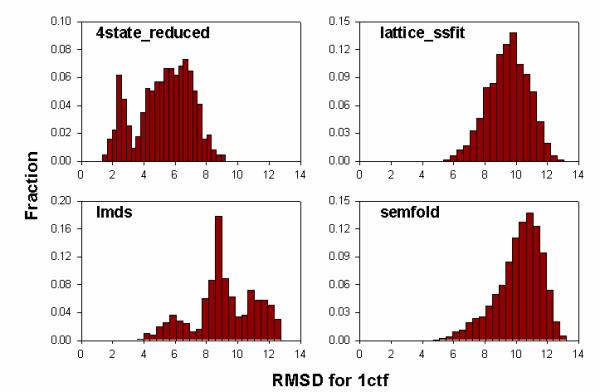
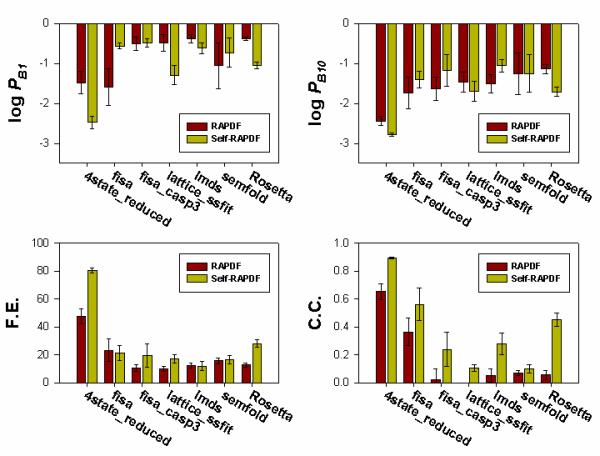
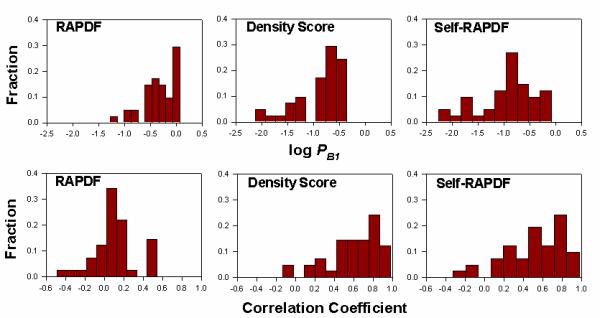
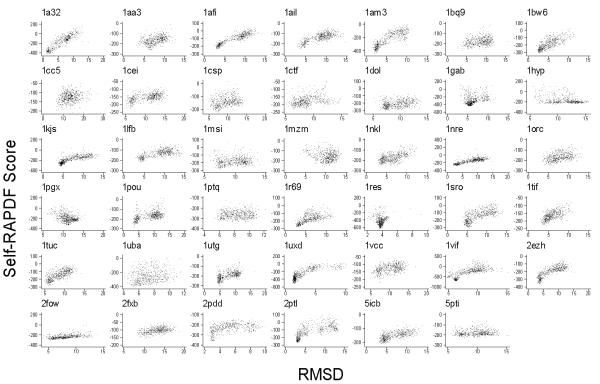
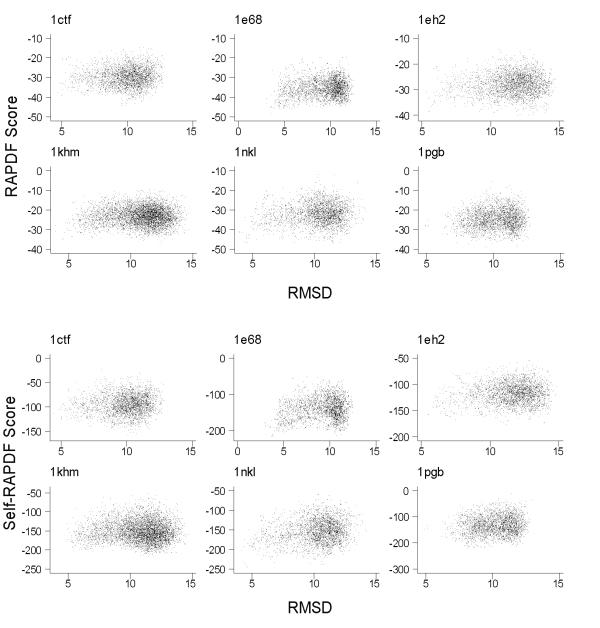
Results: We present a simple and nonparametric formula to estimate the accuracy of predicted conformations (or decoys). This scoring function, called the density score function, evaluates decoy conformations by performing an all-against-all Calpha RMSD (Root Mean Square Deviation) calculation in a given decoy set. We tested the density score function on 83 decoy sets grouped by their generation methods (4state_reduced, fisa, fisa_casp3, lmds, lattice_ssfit, semfold and Rosetta). The density scores have correlations as high as 0.9 with the Calpha RMSDs of the decoy conformations, measured relative to the experimental conformation for each decoy. We previously developed a residue-specific all-atom probability discriminatory function (RAPDF), which compiles statistics from a database of experimentally determined conformations, to aid in structure selection. Here, we present a decoy-dependent discriminatory function called self-RAPDF, where we compiled the atom-atom contact probabilities from all the conformations in a decoy set instead of using an ensemble of native conformations, with a weighting scheme based on the density scores. The self-RAPDF has a higher correlation with Calpha RMSD than RAPDF for 76/83 decoy sets, and selects better near-native conformations for 62/83 decoy sets. Self-RAPDF may be useful not only for selecting near-native conformations from decoy sets, but also for fold simulations and protein structure refinement.
Conclusions: Both the density score and the self-RAPDF functions are decoy-dependent scoring functions for improved protein structure selection. Their success indicates that information from the ensemble of decoy conformations can be used to derive statistical probabilities and facilitate the identification of near-native structures.
Figures

Similar articles
-
Protein structure prediction by all-atom free-energy refinement.BMC Struct Biol. 2007 Mar 19;7:12. doi: 10.1186/1472-6807-7-12. BMC Struct Biol. 2007. PMID: 17371594 Free PMC article.
-
The effect of experimental resolution on the performance of knowledge-based discriminatory functions for protein structure selection.Protein Eng Des Sel. 2006 Sep;19(9):431-7. doi: 10.1093/protein/gzl027. Epub 2006 Jul 14. Protein Eng Des Sel. 2006. PMID: 16845128
-
How well can we predict native contacts in proteins based on decoy structures and their energies?Proteins. 2003 Sep 1;52(4):598-608. doi: 10.1002/prot.10444. Proteins. 2003. PMID: 12910459
-
Methods for the Refinement of Protein Structure 3D Models.Int J Mol Sci. 2019 May 9;20(9):2301. doi: 10.3390/ijms20092301. Int J Mol Sci. 2019. PMID: 31075942 Free PMC article. Review.
-
Advanced Methods for Accessing Protein Shape-Shifting Present New Therapeutic Opportunities.Trends Biochem Sci. 2019 Apr;44(4):351-364. doi: 10.1016/j.tibs.2018.11.007. Epub 2018 Dec 14. Trends Biochem Sci. 2019. PMID: 30555007 Free PMC article. Review.
Cited by
-
Nonbonded terms extrapolated from nonlocal knowledge-based energy functions improve error detection in near-native protein structure models.Protein Sci. 2007 Jul;16(7):1410-21. doi: 10.1110/ps.062735907. Protein Sci. 2007. PMID: 17586774 Free PMC article.
-
Artefacts and biases affecting the evaluation of scoring functions on decoy sets for protein structure prediction.Bioinformatics. 2009 May 15;25(10):1271-9. doi: 10.1093/bioinformatics/btp150. Epub 2009 Mar 17. Bioinformatics. 2009. PMID: 19297350 Free PMC article.
-
A knowledge-based structure-discriminating function that requires only main-chain atom coordinates.BMC Struct Biol. 2008 Oct 29;8:46. doi: 10.1186/1472-6807-8-46. BMC Struct Biol. 2008. PMID: 18957132 Free PMC article.
-
Statistical potential for modeling and ranking of protein-ligand interactions.J Chem Inf Model. 2011 Dec 27;51(12):3078-92. doi: 10.1021/ci200377u. Epub 2011 Nov 21. J Chem Inf Model. 2011. PMID: 22014038 Free PMC article.
-
Statistical potential for assessment and prediction of protein structures.Protein Sci. 2006 Nov;15(11):2507-24. doi: 10.1110/ps.062416606. Protein Sci. 2006. PMID: 17075131 Free PMC article.
References
-
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4:187–217.
-
- Jorgensen William L., Tirado-Rives Julian. The OPLS potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110:1657–1666. - PubMed
-
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197.
-
- Holm L, Sander C. Evaluation of protein models by atomic solvation preference. J Mol Biol. 1992;225:93–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
