

Boosting accuracy of automated classification of fluorescence microscope images for location proteomics
- PMID: 15207009
- PMCID: PMC449699
- DOI: 10.1186/1471-2105-5-78
Boosting accuracy of automated classification of fluorescence microscope images for location proteomics
Abstract
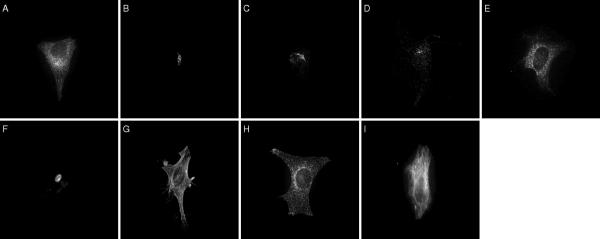
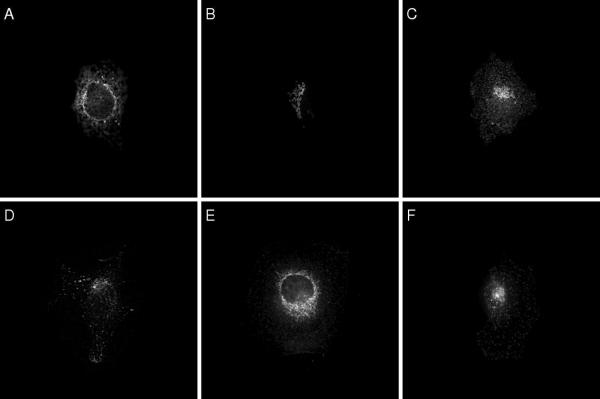
Background: Detailed knowledge of the subcellular location of each expressed protein is critical to a full understanding of its function. Fluorescence microscopy, in combination with methods for fluorescent tagging, is the most suitable current method for proteome-wide determination of subcellular location. Previous work has shown that neural network classifiers can distinguish all major protein subcellular location patterns in both 2D and 3D fluorescence microscope images. Building on these results, we evaluate here new classifiers and features to improve the recognition of protein subcellular location patterns in both 2D and 3D fluorescence microscope images.
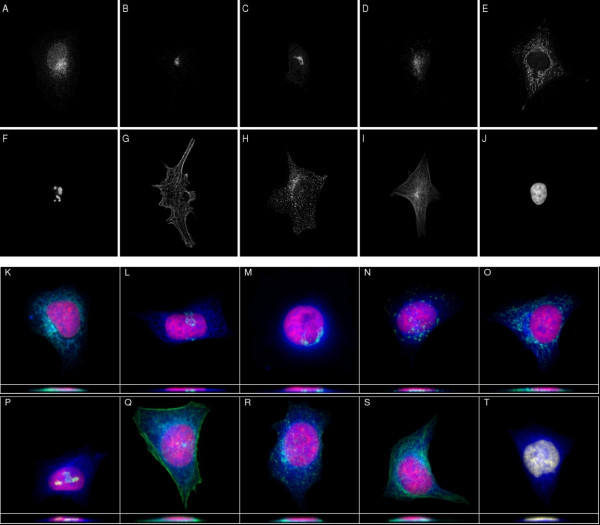
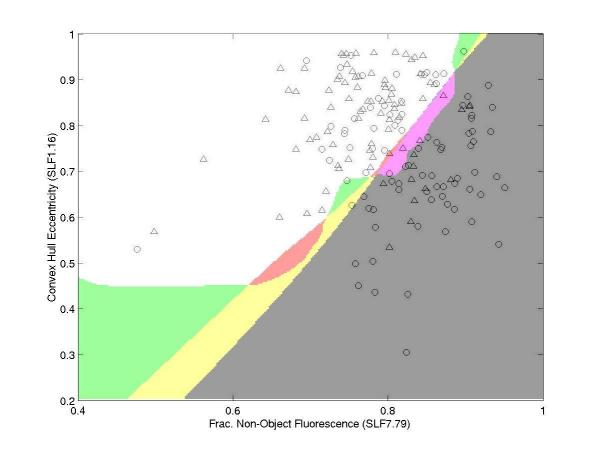
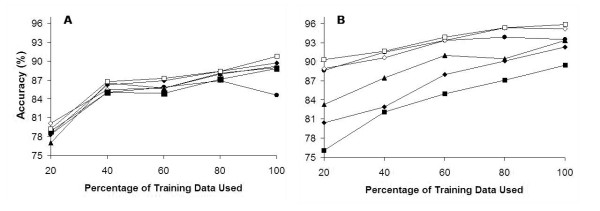
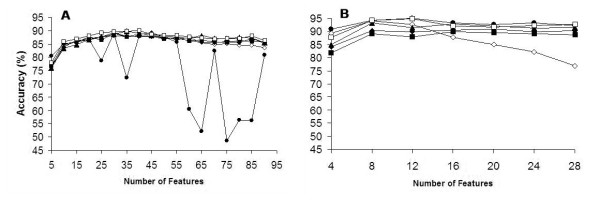
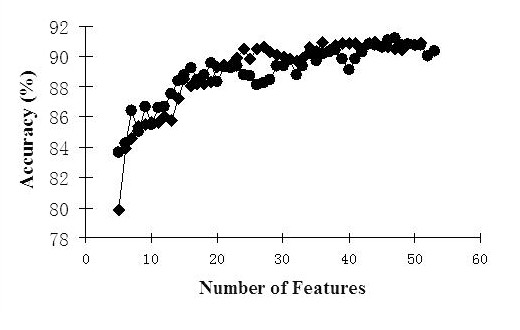
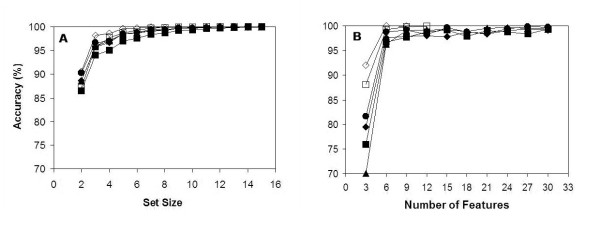
Results: We report here a thorough comparison of the performance on this problem of eight different state-of-the-art classification methods, including neural networks, support vector machines with linear, polynomial, radial basis, and exponential radial basis kernel functions, and ensemble methods such as AdaBoost, Bagging, and Mixtures-of-Experts. Ten-fold cross validation was used to evaluate each classifier with various parameters on different Subcellular Location Feature sets representing both 2D and 3D fluorescence microscope images, including new feature sets incorporating features derived from Gabor and Daubechies wavelet transforms. After optimal parameters were chosen for each of the eight classifiers, optimal majority-voting ensemble classifiers were formed for each feature set. Comparison of results for each image for all eight classifiers permits estimation of the lower bound classification error rate for each subcellular pattern, which we interpret to reflect the fraction of cells whose patterns are distorted by mitosis, cell death or acquisition errors. Overall, we obtained statistically significant improvements in classification accuracy over the best previously published results, with the overall error rate being reduced by one-third to one-half and with the average accuracy for single 2D images being higher than 90% for the first time. In particular, the classification accuracy for the easily confused endomembrane compartments (endoplasmic reticulum, Golgi, endosomes, lysosomes) was improved by 5-15%. We achieved further improvements when classification was conducted on image sets rather than on individual cell images.
Conclusions: The availability of accurate, fast, automated classification systems for protein location patterns in conjunction with high throughput fluorescence microscope imaging techniques enables a new subfield of proteomics, location proteomics. The accuracy and sensitivity of this approach represents an important alternative to low-resolution assignments by curation or sequence-based prediction.
Figures
Similar articles
-
Phenotype recognition with combined features and random subspace classifier ensemble.BMC Bioinformatics. 2011 Apr 30;12:128. doi: 10.1186/1471-2105-12-128. BMC Bioinformatics. 2011. PMID: 21529372 Free PMC article.
-
A graphical model approach to automated classification of protein subcellular location patterns in multi-cell images.BMC Bioinformatics. 2006 Feb 23;7:90. doi: 10.1186/1471-2105-7-90. BMC Bioinformatics. 2006. PMID: 16504075 Free PMC article.
-
A multiresolution approach to automated classification of protein subcellular location images.BMC Bioinformatics. 2007 Jun 19;8:210. doi: 10.1186/1471-2105-8-210. BMC Bioinformatics. 2007. PMID: 17578580 Free PMC article.
-
Automated interpretation of subcellular patterns in fluorescence microscope images for location proteomics.Cytometry A. 2006 Jul;69(7):631-40. doi: 10.1002/cyto.a.20280. Cytometry A. 2006. PMID: 16752421 Free PMC article. Review.
-
Automated, systematic determination of protein subcellular location using fluorescence microscopy.Subcell Biochem. 2007;43:263-76. doi: 10.1007/978-1-4020-5943-8_12. Subcell Biochem. 2007. PMID: 17953398 Review.
Cited by
-
Data-mining Techniques for Image-based Plant Phenotypic Traits Identification and Classification.Sci Rep. 2019 Dec 20;9(1):19526. doi: 10.1038/s41598-019-55609-6. Sci Rep. 2019. PMID: 31862925 Free PMC article.
-
Large-scale automated analysis of location patterns in randomly tagged 3T3 cells.Ann Biomed Eng. 2007 Jun;35(6):1081-7. doi: 10.1007/s10439-007-9254-5. Epub 2007 Feb 7. Ann Biomed Eng. 2007. PMID: 17285363 Free PMC article.
-
The Open Microscopy Environment (OME) Data Model and XML file: open tools for informatics and quantitative analysis in biological imaging.Genome Biol. 2005;6(5):R47. doi: 10.1186/gb-2005-6-5-r47. Epub 2005 May 3. Genome Biol. 2005. PMID: 15892875 Free PMC article.
-
Objective clustering of proteins based on subcellular location patterns.J Biomed Biotechnol. 2005 Jun 30;2005(2):87-95. doi: 10.1155/JBB.2005.87. J Biomed Biotechnol. 2005. PMID: 16046813 Free PMC article.
-
Determining the subcellular location of new proteins from microscope images using local features.Bioinformatics. 2013 Sep 15;29(18):2343-9. doi: 10.1093/bioinformatics/btt392. Epub 2013 Jul 8. Bioinformatics. 2013. PMID: 23836142 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
