

Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus
- PMID: 15208709
- PMCID: PMC423131
- DOI: 10.1371/journal.pbio.0020142
Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus
Abstract
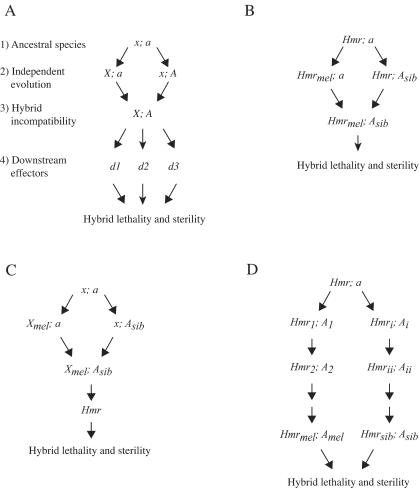
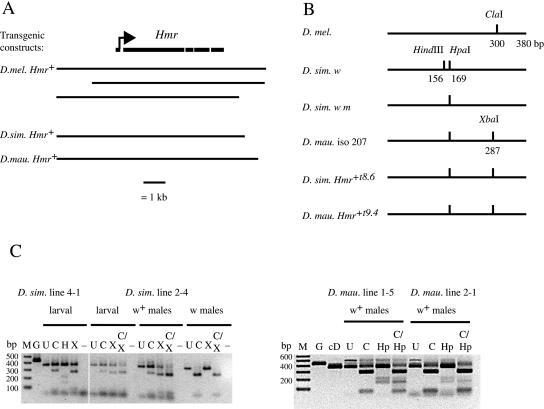
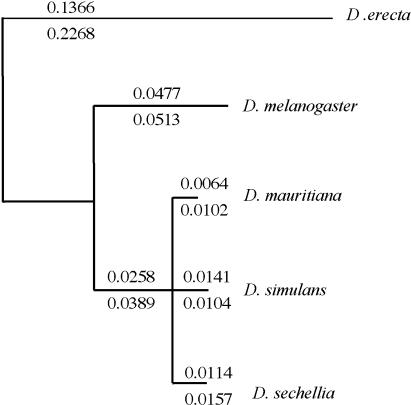
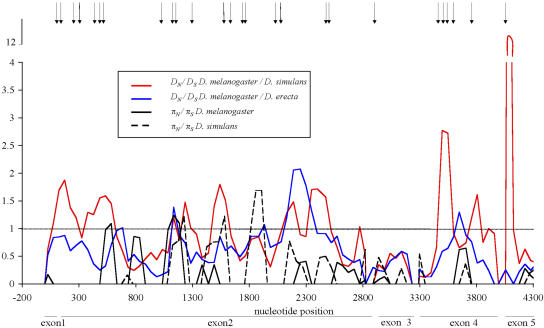
Interspecific hybrid lethality and sterility are a consequence of divergent evolution between species and serve to maintain the discrete identities of species. The evolution of hybrid incompatibilities has been described in widely accepted models by Dobzhansky and Muller where lineage-specific functional divergence is the essential characteristic of hybrid incompatibility genes. Experimentally tractable models are required to identify and test candidate hybrid incompatibility genes. Several Drosophila melanogaster genes involved in hybrid incompatibility have been identified but none has yet been shown to have functionally diverged in accordance with the Dobzhansky-Muller model. By introducing transgenic copies of the X-linked Hybrid male rescue (Hmr) gene into D. melanogaster from its sibling species D. simulans and D. mauritiana, we demonstrate that Hmr has functionally diverged to cause F1 hybrid incompatibility between these species. Consistent with the Dobzhansky-Muller model, we find that Hmr has diverged extensively in the D. melanogaster lineage, but we also find extensive divergence in the sibling-species lineage. Together, these findings implicate over 13% of the amino acids encoded by Hmr as candidates for causing hybrid incompatibility. The exceptional level of divergence at Hmr cannot be explained by neutral processes because we use phylogenetic methods and population genetic analyses to show that the elevated amino-acid divergence in both lineages is due to positive selection in the distant past-at least one million generations ago. Our findings suggest that multiple substitutions driven by natural selection may be a general phenomenon required to generate hybrid incompatibility alleles.
Conflict of interest statement
The authors have declared that no conflicts of interest exist.
Figures

References
-
- Andolfatto P. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans . Mol Biol Evol. 2001;18:279–290. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
