

The CHAOS/DIALIGN WWW server for multiple alignment of genomic sequences
- PMID: 15215346
- PMCID: PMC441499
- DOI: 10.1093/nar/gkh361
The CHAOS/DIALIGN WWW server for multiple alignment of genomic sequences
Abstract
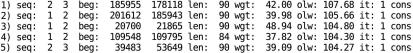
Cross-species sequence comparison is a powerful approach to analyze functional sites in genomic sequences and many discoveries have been made based on genomic alignments. Herein, we present a WWW-based software system for multiple alignment of large genomic sequences. Our server utilizes the previously developed combination of CHAOS and DIALIGN to achieve both speed and alignment accuracy. CHAOS is a fast database search tool that creates a list of local sequence similarities. These are used by DIALIGN as anchor points to speed up the final alignment procedure. The resulting alignment is returned to the user in different formats together with a list of anchor points found by CHAOS. The CHAOS/DIALIGN software is freely available at http://dialign.gobics.de/chaos-dialign-submission.
Figures

References
-
- Bafna V. and Huson,D.H. (2000) The conserved exon method for gene finding. Bioinformatics, 16, 190–202. - PubMed
-
- Korf I., Flicek,P., Duan,D. and Brent,M.R. (2001) Integrating genomic homology into gene structure prediction. Bioinformatics, 17, S140–S148. - PubMed
-
- Taher L., Rinner,O., Gargh,S., Sczyrba,A., Brudno,M., Batzoglou,S. and Morgenstern,B. (2003) AGenDA: homology-based gene prediction. Bioinformatics, 19, 1575–1577. - PubMed
