

Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information
- PMID: 15215532
- PMCID: PMC2279939
- DOI: 10.1110/ps.04625404
Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information
Abstract
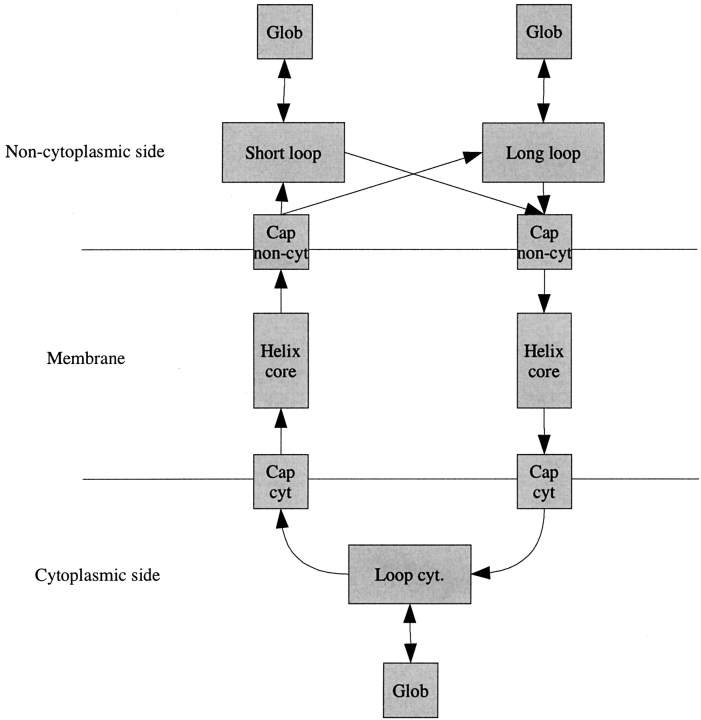
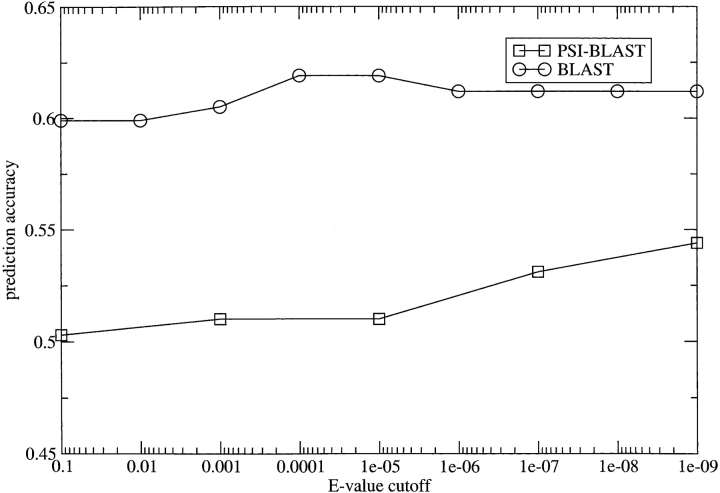
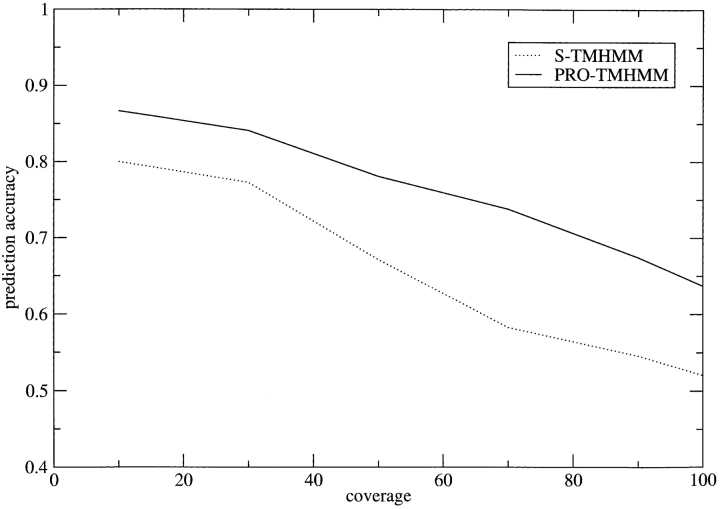
Methods that predict the topology of helical membrane proteins are standard tools when analyzing any proteome. Therefore, it is important to improve the performance of such methods. Here we introduce a novel method, PRODIV-TMHMM, which is a profile-based hidden Markov model (HMM) that also incorporates the best features of earlier HMM methods. In our tests, PRODIV-TMHMM outperforms earlier methods both when evaluated on "low-resolution" topology data and on high-resolution 3D structures. The results presented here indicate that the topology could be correctly predicted for approximately two-thirds of all membrane proteins using PRODIV-TMHMM. The importance of evolutionary information for topology prediction is emphasized by the fact that compared with using single sequences, the performance of PRODIV-TMHMM (as well as two other methods) is increased by approximately 10 percentage units by the use of homologous sequences. On a more general level, we also show that HMM-based (or similar) methods perform superiorly to methods that focus mainly on identification of the membrane regions.
Figures
References
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. - PubMed
-
- Claros, M.G. and von Heijne, G. 1994. Toppred II: An improved software for membrane protein structure prediction. Comput. Appl. Biosci. 10 685–686. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
