

Joint analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes
- PMID: 15217521
- PMCID: PMC476733
- DOI: 10.1186/1471-2105-5-81
Joint analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes
Abstract
Background: Due to the high cost and low reproducibility of many microarray experiments, it is not surprising to find a limited number of patient samples in each study, and very few common identified marker genes among different studies involving patients with the same disease. Therefore, it is of great interest and challenge to merge data sets from multiple studies to increase the sample size, which may in turn increase the power of statistical inferences. In this study, we combined two lung cancer studies using microarray GeneChip, employed two gene shaving methods and a two-step survival test to identify genes with expression patterns that can distinguish diseased from normal samples, and to indicate patient survival, respectively.
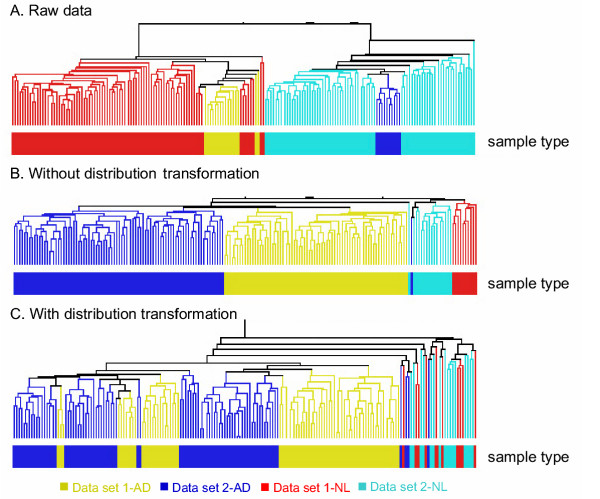
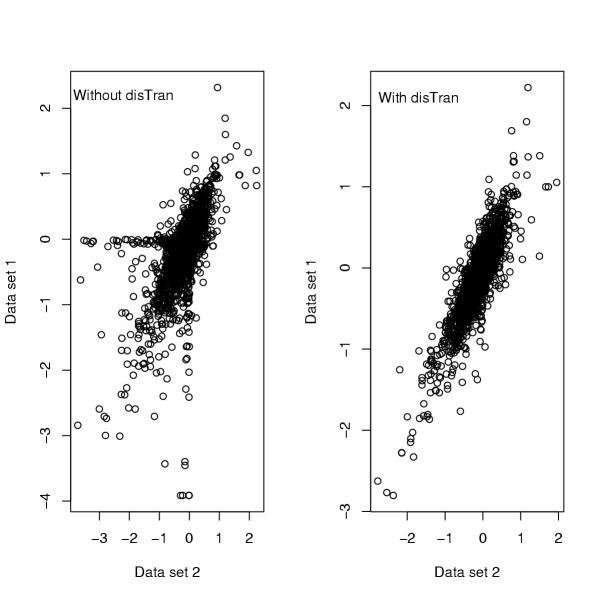
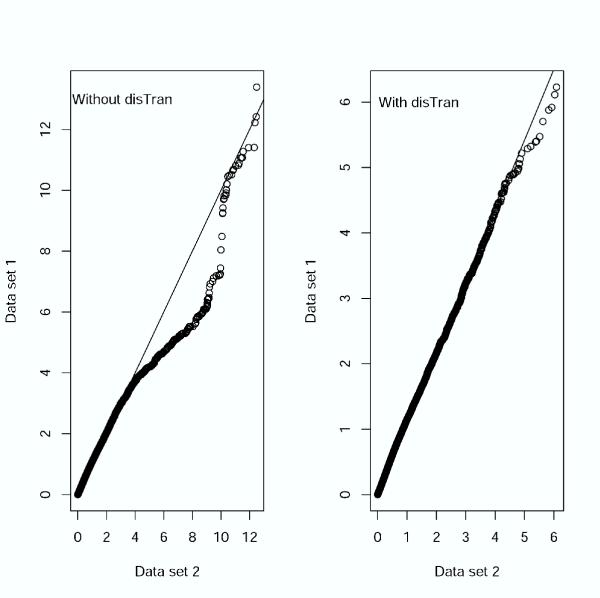
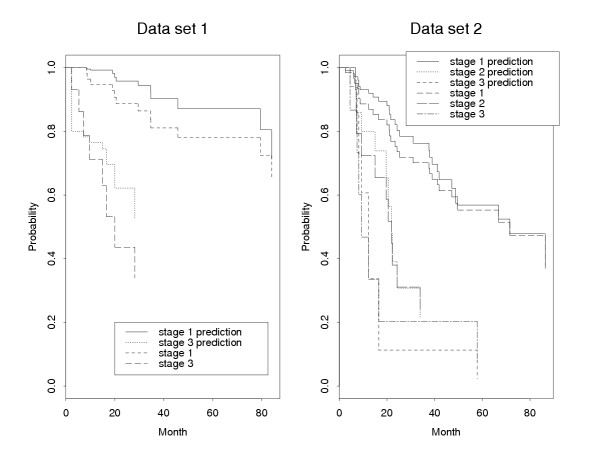
Results: In addition to common data transformation and normalization procedures, we applied a distribution transformation method to integrate the two data sets. Gene shaving (GS) methods based on Random Forests (RF) and Fisher's Linear Discrimination (FLD) were then applied separately to the joint data set for cancer gene selection. The two methods discovered 13 and 10 marker genes (5 in common), respectively, with expression patterns differentiating diseased from normal samples. Among these marker genes, 8 and 7 were found to be cancer-related in other published reports. Furthermore, based on these marker genes, the classifiers we built from one data set predicted the other data set with more than 98% accuracy. Using the univariate Cox proportional hazard regression model, the expression patterns of 36 genes were found to be significantly correlated with patient survival (p < 0.05). Twenty-six of these 36 genes were reported as survival-related genes from the literature, including 7 known tumor-suppressor genes and 9 oncogenes. Additional principal component regression analysis further reduced the gene list from 36 to 16.
Conclusion: This study provided a valuable method of integrating microarray data sets with different origins, and new methods of selecting a minimum number of marker genes to aid in cancer diagnosis. After careful data integration, the classification method developed from one data set can be applied to the other with high prediction accuracy.
Figures
References
-
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. - PubMed
-
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. - DOI - PMC - PubMed
-
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
