

Glycine protects cardiomyocytes against lethal reoxygenation injury by inhibiting mitochondrial permeability transition
- PMID: 15218075
- PMCID: PMC1665014
- DOI: 10.1113/jphysiol.2004.068320
Glycine protects cardiomyocytes against lethal reoxygenation injury by inhibiting mitochondrial permeability transition
Abstract
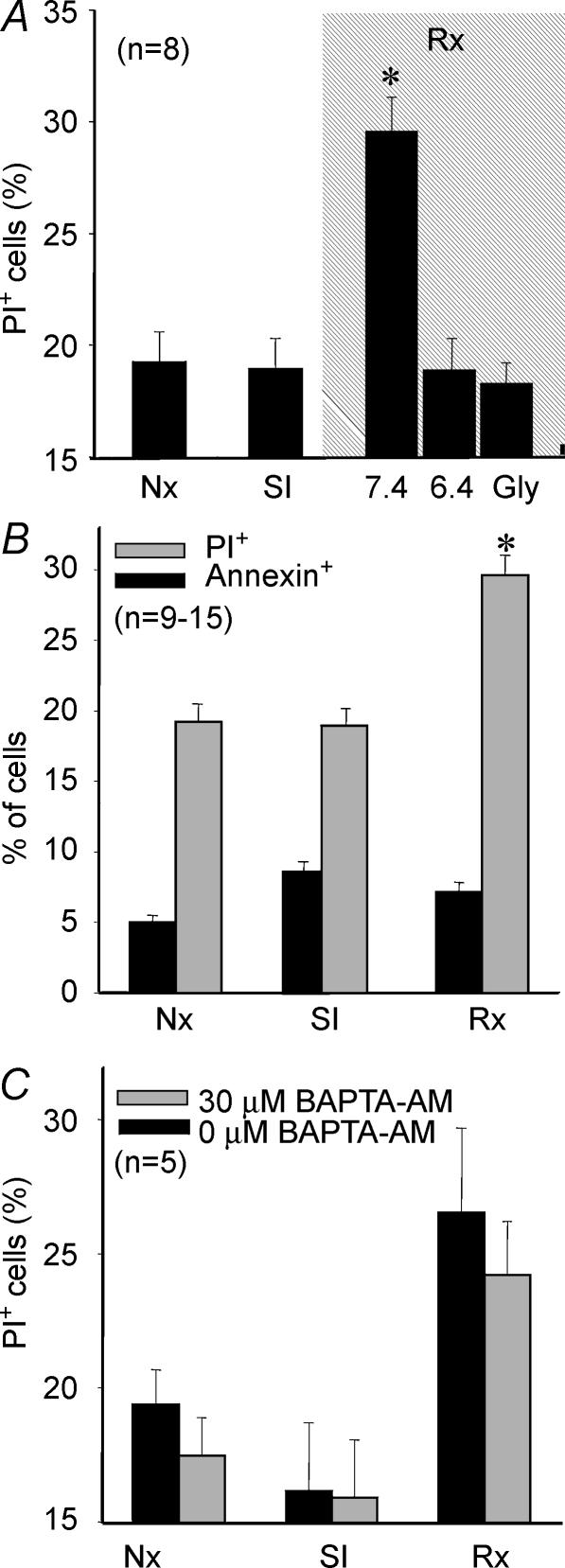
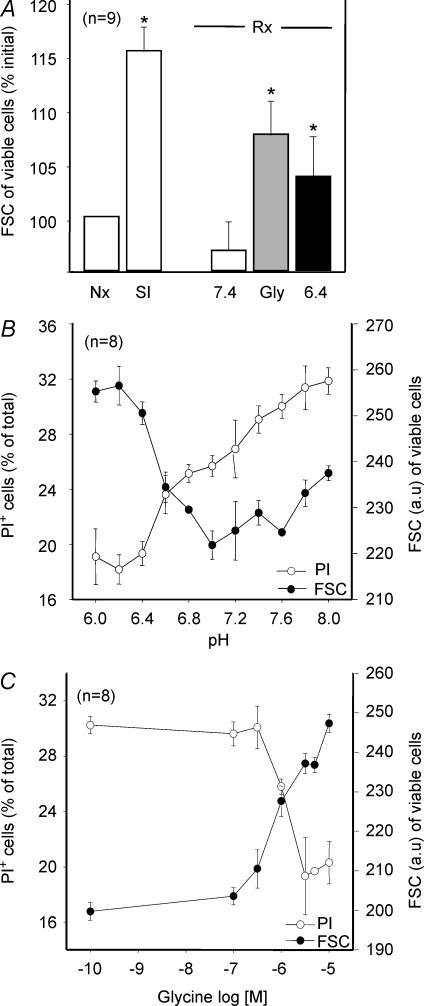
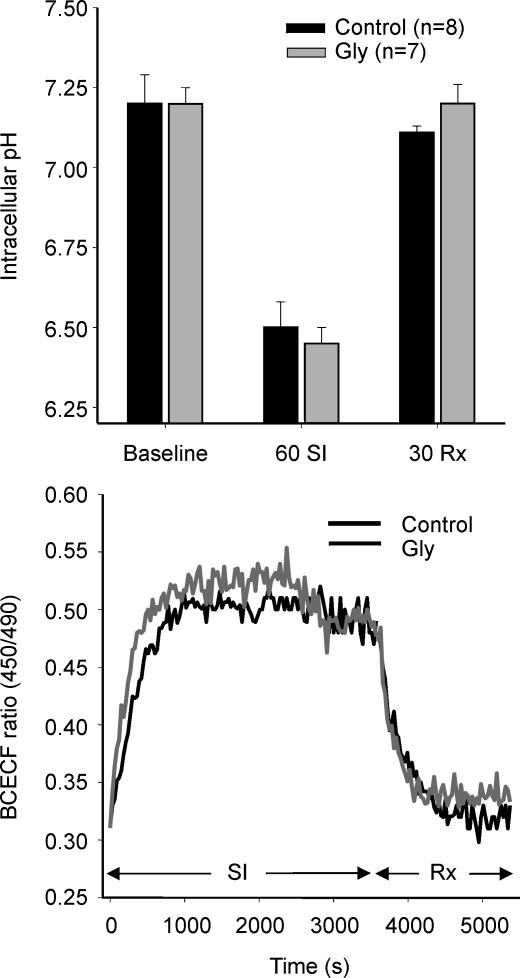
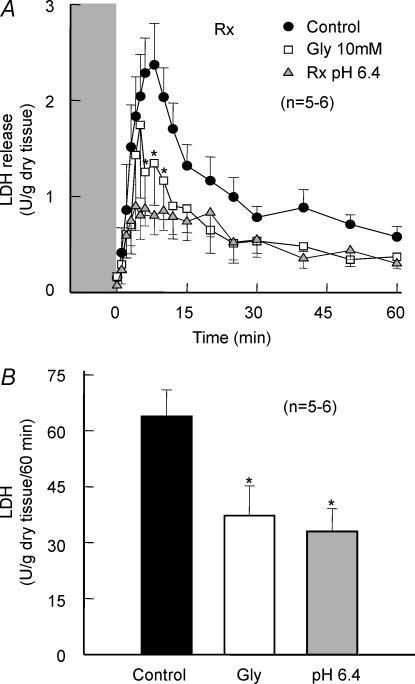
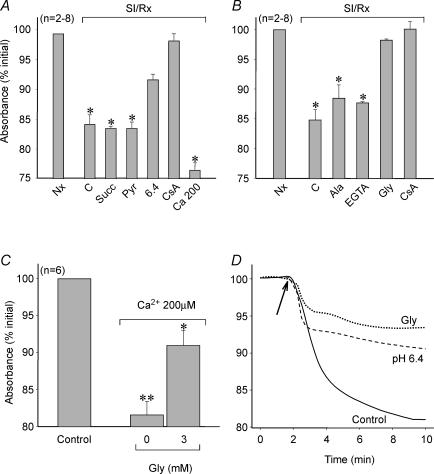
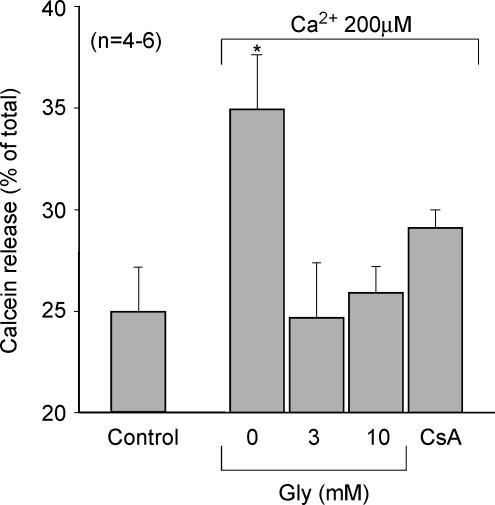
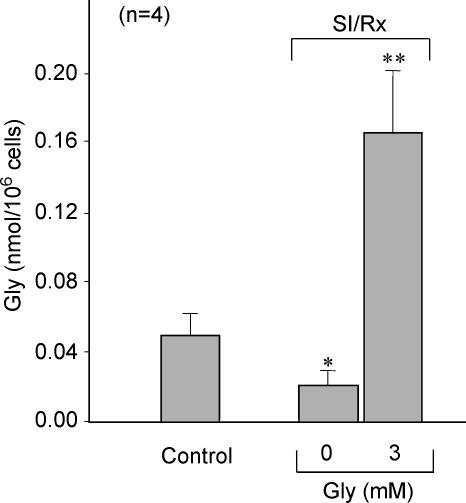
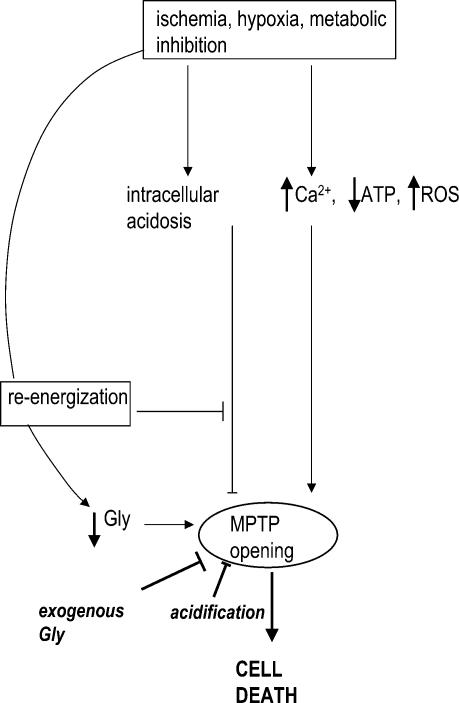
Post-ischaemic reperfusion may precipitate cardiomyocyte death upon correction of intracellular acidosis due in part to mitochondrial permeability transition. We investigated whether glycine, an amino acid with poorly understood cytoprotective properties, may interfere with this mechanism. In cardiomyocyte cultures, addition of glycine during re-energization following 1 h of simulated ischaemia (NaCN/2-deoxyglucose, pH 6.4) completely prevented necrotic cell death associated with pH normalization. Glycine also protected against cell death associated with pH normalization in reoxygenated rat hearts. Glycine prevented cyclosporin-sensitive swelling and calcein release associated with re-energization in rat heart mitochondria submitted to simulated ischaemia or to Ca(2+) stress under normoxia. NMR spectroscopy revealed a marked glycine depletion in re-energized cardiomyocytes that was reversed by exposure to 3 mm glycine. These results suggest that intracellular glycine exerts a previously unrecognized inhibition on mitochondrial permeability transition in cardiac myocytes, and that intracellular glycine depletion during myocardial hypoxia/reoxygenation makes the cell more vulnerable to necrotic death.
Figures
Similar articles
-
Evidence that hydroxysafflor yellow A protects the heart against ischaemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening.Clin Exp Pharmacol Physiol. 2008 Feb;35(2):211-6. doi: 10.1111/j.1440-1681.2007.04814.x. Epub 2007 Oct 17. Clin Exp Pharmacol Physiol. 2008. PMID: 17941891
-
Enhanced cell-volume regulation in cyclosporin A cardioprotection.Cardiovasc Res. 2013 Jun 1;98(3):411-9. doi: 10.1093/cvr/cvt056. Epub 2013 Mar 12. Cardiovasc Res. 2013. PMID: 23483048
-
Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury.Circulation. 2013 Oct 1;128(14):1555-65. doi: 10.1161/CIRCULATIONAHA.113.001225. Epub 2013 Aug 27. Circulation. 2013. PMID: 23983249
-
CXCR4 attenuates cardiomyocytes mitochondrial dysfunction to resist ischaemia-reperfusion injury.J Cell Mol Med. 2015 Aug;19(8):1825-35. doi: 10.1111/jcmm.12554. Epub 2015 Mar 30. J Cell Mol Med. 2015. PMID: 25824297 Free PMC article.
-
The role of glycine in regulated cell death.Cell Mol Life Sci. 2016 Jun;73(11-12):2285-308. doi: 10.1007/s00018-016-2201-6. Epub 2016 Apr 11. Cell Mol Life Sci. 2016. PMID: 27066896 Free PMC article. Review.
Cited by
-
Circulating mediators of remote ischemic preconditioning: search for the missing link between non-lethal ischemia and cardioprotection.Oncotarget. 2019 Jan 4;10(2):216-244. doi: 10.18632/oncotarget.26537. eCollection 2019 Jan 4. Oncotarget. 2019. PMID: 30719216 Free PMC article. Review.
-
Glycine attenuates myocardial ischemia-reperfusion injury by inhibiting myocardial apoptosis in rats.J Biomed Res. 2012 Sep;26(5):346-54. doi: 10.7555/JBR.26.20110124. Epub 2012 Jun 29. J Biomed Res. 2012. PMID: 23554770 Free PMC article.
-
Metabolic changes in mice cardiac tissue after low-dose irradiation revealed by 1H NMR spectroscopy.J Radiat Res. 2020 Jan 23;61(1):14-26. doi: 10.1093/jrr/rrz079. J Radiat Res. 2020. PMID: 31840756 Free PMC article.
-
Glycine inhibits the LPS-induced increase in cytosolic Ca2+ concentration and TNFalpha production in cardiomyocytes by activating a glycine receptor.Acta Pharmacol Sin. 2009 Aug;30(8):1107-14. doi: 10.1038/aps.2009.106. Epub 2009 Jul 20. Acta Pharmacol Sin. 2009. PMID: 19617896 Free PMC article.
-
The cardiometabolic benefits of glycine: Is glycine an 'antidote' to dietary fructose?Open Heart. 2014 May 28;1(1):e000103. doi: 10.1136/openhrt-2014-000103. eCollection 2014. Open Heart. 2014. PMID: 25332814 Free PMC article. No abstract available.
References
-
- Carini R, De Cesaris MG, Splendore R, Bagnati M, Bellomo G, Albano E. Alterations of Na+ homeostasis in hepatocyte reoxygenation injury. Biochim Biophys Acta. 2000;1500:297–305. - PubMed
-
- De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ, Ichas F. The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J. 2002;16:607–609. - PubMed
-
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. - PubMed
-
- Dong Z, Patel Y, Saikumar P, Weinberg JM, Venkatachalam MA. Development of porous defects in plasma membranes of adenosine triphosphate-depleted Madin-Darby canine kidney cells and its inhibition by glycine. Laboratory Invest. 1998;78:657–668. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous