

Regulog analysis: detection of conserved regulatory networks across bacteria: application to Staphylococcus aureus
- PMID: 15231752
- PMCID: PMC442153
- DOI: 10.1101/gr.2242604
Regulog analysis: detection of conserved regulatory networks across bacteria: application to Staphylococcus aureus
Abstract
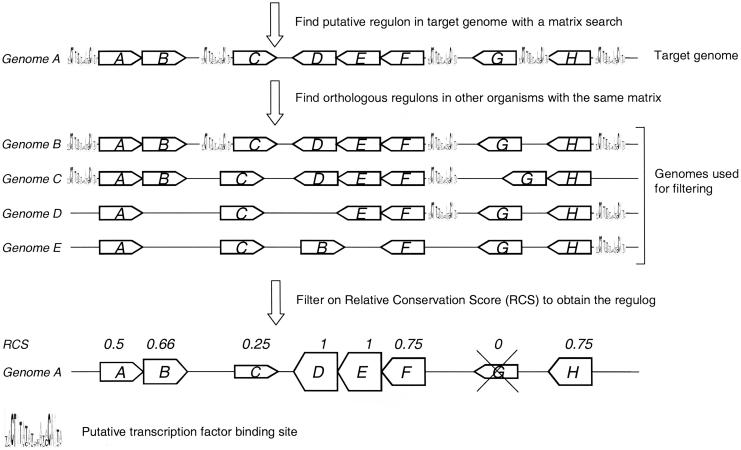
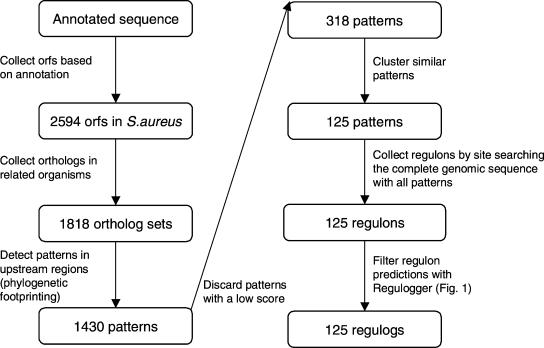
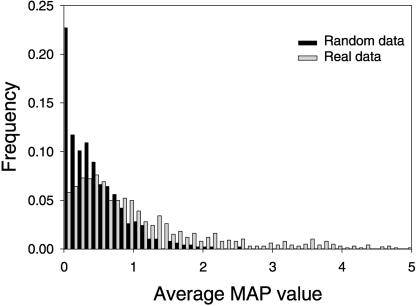
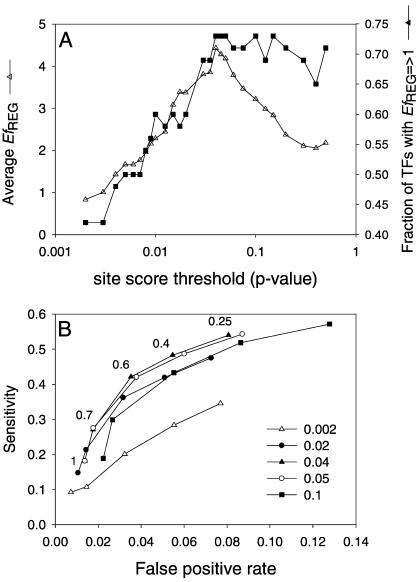
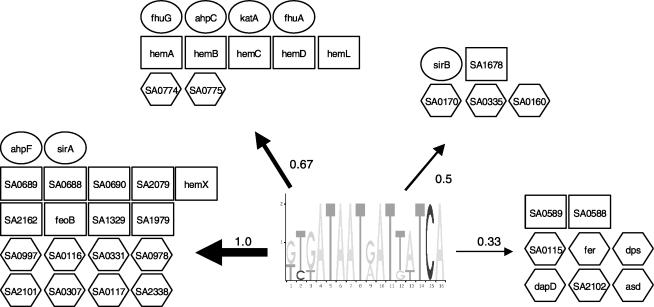
A transcriptional regulatory network encompasses sets of genes (regulons) whose expression states are directly altered in response to an activating signal, mediated by trans-acting regulatory proteins and cis-acting regulatory sequences. Enumeration of these network components is an essential step toward the creation of a framework for systems-based analysis of biological processes. Profile-based methods for the detection of cis-regulatory elements are often applied to predict regulon members, but they suffer from poor specificity. In this report we describe Regulogger, a novel computational method that uses comparative genomics to eliminate spurious members of predicted gene regulons. Regulogger produces regulogs, sets of coregulated genes for which the regulatory sequence has been conserved across multiple organisms. The quantitative method assigns a confidence score to each predicted regulog member on the basis of the degree of conservation of protein sequence and regulatory mechanisms. When applied to a reference collection of regulons from Escherichia coli, Regulogger increased the specificity of predictions up to 25-fold over methods that use cis-element detection in isolation. The enhanced specificity was observed across a wide range of biologically meaningful parameter combinations, indicating a robust and broad utility for the method. The power of computational pattern discovery methods coupled with Regulogger to unravel transcriptional networks was demonstrated in an analysis of the genome of Staphylococcus aureus. A total of 125 regulogs were found in this organism, including both well-defined functional groups and a subset with unknown functions.
Copyright 2004 Cold Spring Harbor Laboratory Press ISSN
Figures
References
-
- Baichoo, N., Wang, T., Ye, R., and Helmann, J.D. 2002. Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol. Microbiol. 45: 1613–1629. - PubMed
-
- Bockhorst, J., Craven, M., Page, D., Shavlik, J., and Glasner, J. 2003. A Bayesian network approach to operon prediction. Bioinformatics 19: 1227–1235. - PubMed
WEB SITE REFERENCES
-
- http://www.ncbi.nlm.nih.gov; NCBI home page.
-
- http://arep.med.harvard.edu/dpinteract; DPInteract.
-
- http://www.weizmann.ac.il/mcb/UriAlon; Alon Lab.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources