

Base-By-Base: single nucleotide-level analysis of whole viral genome alignments
- PMID: 15253776
- PMCID: PMC481056
- DOI: 10.1186/1471-2105-5-96
Base-By-Base: single nucleotide-level analysis of whole viral genome alignments
Abstract
Background: With ever increasing numbers of closely related virus genomes being sequenced, it has become desirable to be able to compare two genomes at a level more detailed than gene content because two strains of an organism may share the same set of predicted genes but still differ in their pathogenicity profiles. For example, detailed comparison of multiple isolates of the smallpox virus genome (each approximately 200 kb, with 200 genes) is not feasible without new bioinformatics tools.
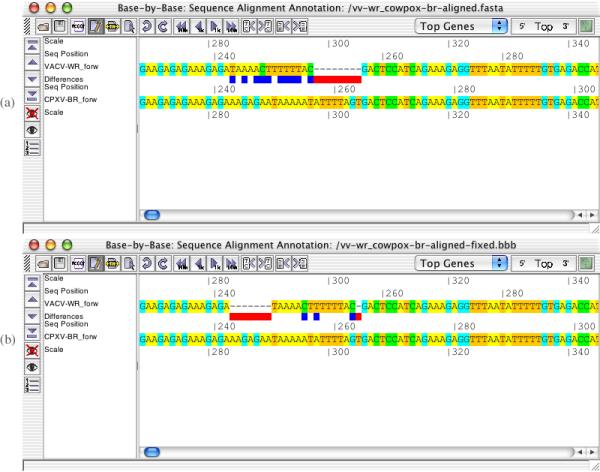
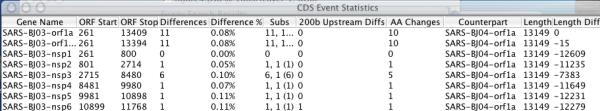
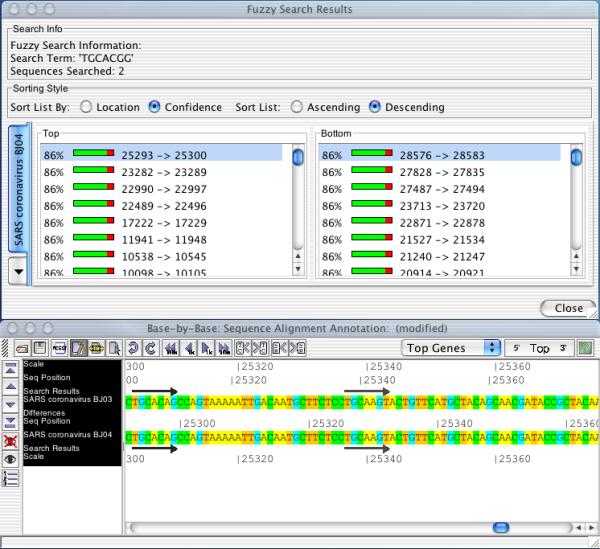
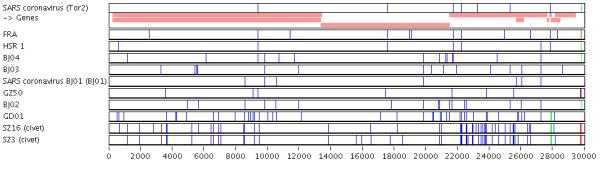
Results: A software package, Base-By-Base, has been developed that provides visualization tools to enable researchers to 1) rapidly identify and correct alignment errors in large, multiple genome alignments; and 2) generate tabular and graphical output of differences between the genomes at the nucleotide level. Base-By-Base uses detailed annotation information about the aligned genomes and can list each predicted gene with nucleotide differences, display whether variations occur within promoter regions or coding regions and whether these changes result in amino acid substitutions. Base-By-Base can connect to our mySQL database (Virus Orthologous Clusters; VOCs) to retrieve detailed annotation information about the aligned genomes or use information from text files.
Conclusion: Base-By-Base enables users to quickly and easily compare large viral genomes; it highlights small differences that may be responsible for important phenotypic differences such as virulence. It is available via the Internet using Java Web Start and runs on Macintosh, PC and Linux operating systems with the Java 1.4 virtual machine.
Figures

References
-
- Moss B. Poxviruses. In: Knipe DM and Howley P M, editor. Fields Virology. Vol. 2. Philadelphia, Lippincott Williams & Wilkins; 2001. pp. 2849–2884.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
