

In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation
- PMID: 15254240
- PMCID: PMC444865
- DOI: 10.1128/MCB.24.15.6728-6741.2004
In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation
Abstract
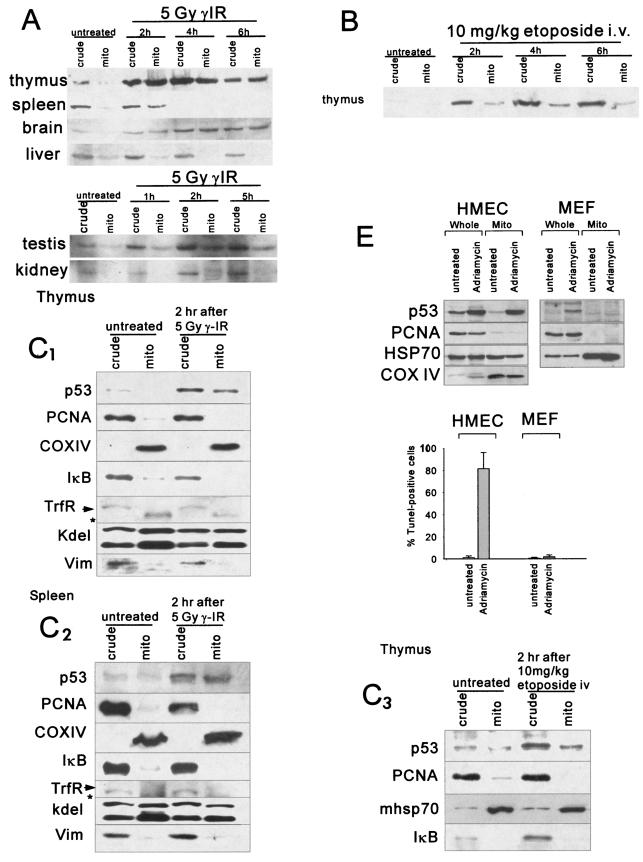
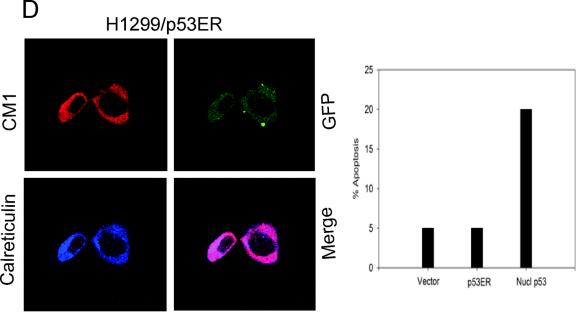
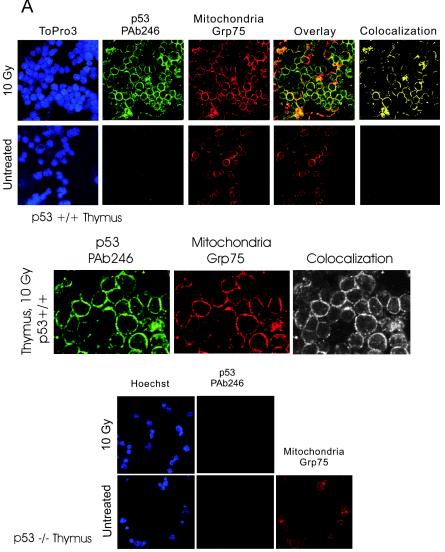
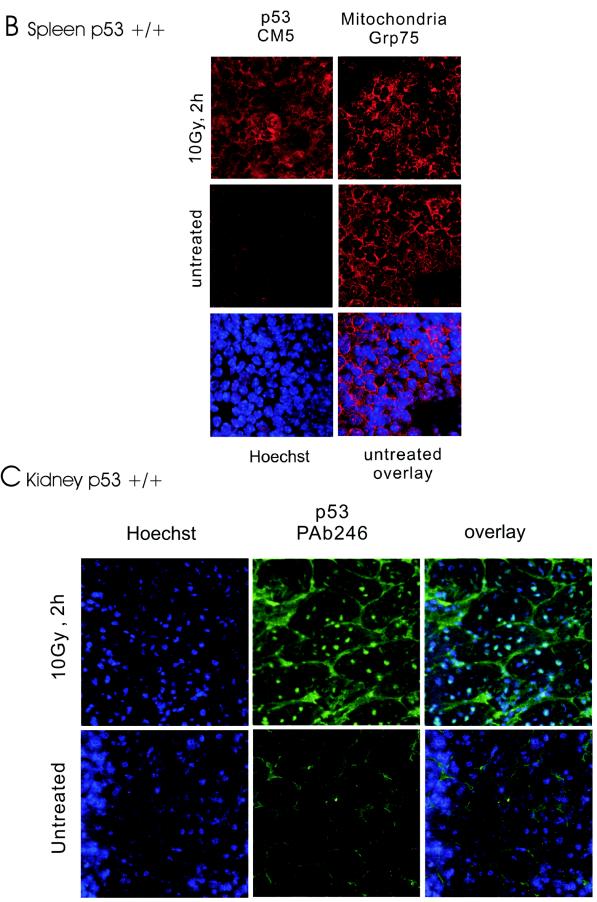
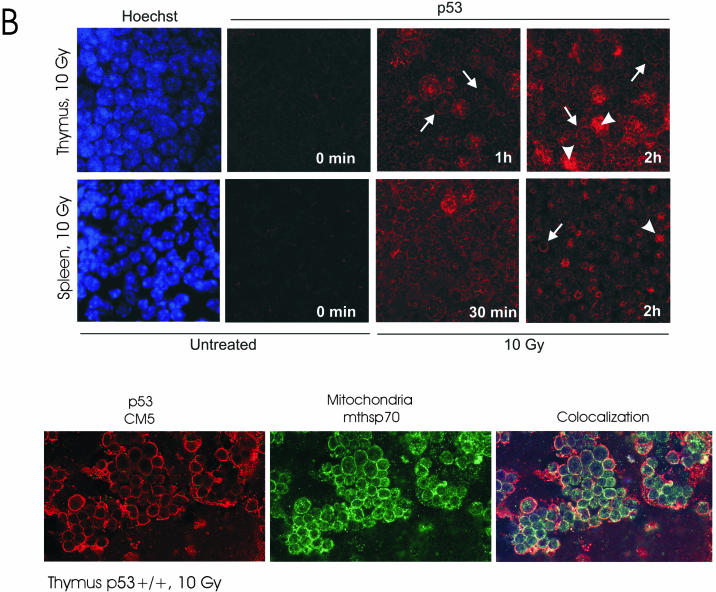
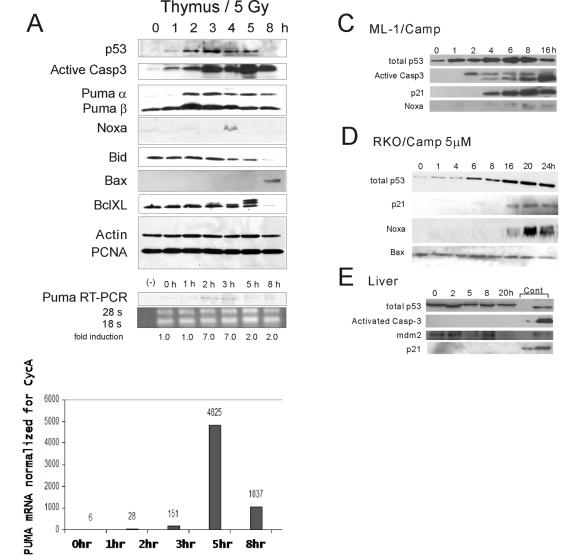
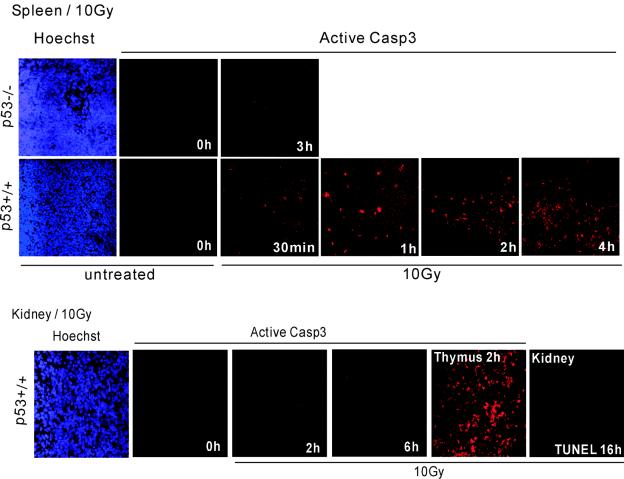
p53 promotes apoptosis in response to death stimuli by transactivation of target genes and by transcription-independent mechanisms. We recently showed that wild-type p53 rapidly translocates to mitochondria in response to multiple death stimuli in cultured cells. Mitochondrial p53 physically interacts with antiapoptotic Bcl proteins, induces Bak oligomerization, permeabilizes mitochondrial membranes, and rapidly induces cytochrome c release. Here we characterize the mitochondrial p53 response in vivo. Mice were subjected to gamma irradiation or intravenous etoposide administration, followed by cell fractionation and immunofluorescence studies of various organs. Mitochondrial p53 accumulation occurred in radiosensitive organs like thymus, spleen, testis, and brain but not in liver and kidney. Of note, mitochondrial p53 translocation was rapid (detectable at 30 min in thymus and spleen) and triggered an early wave of marked caspase 3 activation and apoptosis. This caspase 3-mediated apoptosis was entirely p53 dependent, as shown by p53 null mice, and preceded p53 target gene activation. The transcriptional p53 program had a longer lag phase than the rapid mitochondrial p53 program. In thymus, the earliest apoptotic target gene products PUMA, Noxa, and Bax appeared at 2, 4, and 8 h, respectively, while Bid, Killer/DR5, and p53DinP1 remained uninduced even after 20 h. Target gene induction then led to further increase in active caspase 3. Similar biphasic kinetics was seen in cultured human cells. Our results suggest that in sensitive organs mitochondrial p53 accumulation in vivo occurs soon after a death stimulus, triggering a rapid first wave of apoptosis that is transcription independent and may precede a second slower wave that is transcription dependent.
Figures


Similar articles
-
Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo.Oncogene. 2001 Aug 2;20(34):4601-12. doi: 10.1038/sj.onc.1204484. Oncogene. 2001. PMID: 11498783
-
Tissue-specific induction of p53 targets in vivo.Cancer Res. 2002 Dec 15;62(24):7316-27. Cancer Res. 2002. PMID: 12499275
-
A comprehensive study of p53 transcriptional activity in thymus and spleen of gamma irradiated mouse: high sensitivity of genes involved in the two main apoptotic pathways.Int J Radiat Biol. 2006 Nov;82(11):761-70. doi: 10.1080/09553000600949624. Int J Radiat Biol. 2006. PMID: 17148260
-
Mechanisms of p53-dependent apoptosis.Biochem Soc Trans. 2001 Nov;29(Pt 6):684-8. doi: 10.1042/0300-5127:0290684. Biochem Soc Trans. 2001. PMID: 11709054 Review.
-
Mitochondrial-nuclear p53 trafficking controls neuronal susceptibility in stroke.IUBMB Life. 2021 Mar;73(3):582-591. doi: 10.1002/iub.2453. Epub 2021 Mar 2. IUBMB Life. 2021. PMID: 33615665 Free PMC article. Review.
Cited by
-
Engineered adenovirus fiber shaft fusion homotrimer of soluble TRAIL with enhanced stability and antitumor activity.Cell Death Dis. 2016 Jun 23;7(6):e2274. doi: 10.1038/cddis.2016.177. Cell Death Dis. 2016. PMID: 27336718 Free PMC article.
-
Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite.Mol Cell Biol. 2005 Oct;25(19):8732-47. doi: 10.1128/MCB.25.19.8732-8747.2005. Mol Cell Biol. 2005. PMID: 16166651 Free PMC article.
-
Quercetin induced ROS production triggers mitochondrial cell death of human embryonic stem cells.Oncotarget. 2016 Aug 5;8(39):64964-64973. doi: 10.18632/oncotarget.11070. eCollection 2017 Sep 12. Oncotarget. 2016. PMID: 29029404 Free PMC article.
-
SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization.Cell Stem Cell. 2008 Mar 6;2(3):241-51. doi: 10.1016/j.stem.2008.01.002. Cell Stem Cell. 2008. PMID: 18371449 Free PMC article.
-
Modulation of p53, c-fos, RARE, cyclin A, and cyclin D1 expression in human leukemia (HL-60) cells exposed to arsenic trioxide.Mol Cell Biochem. 2009 Nov;331(1-2):207-14. doi: 10.1007/s11010-009-0160-z. Epub 2009 May 15. Mol Cell Biochem. 2009. PMID: 19444595 Free PMC article.
References
-
- Baptiste, N., P. Friedlander, X. Chen, and C. Prives. 2002. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 21:9-21. - PubMed
-
- Bennett, M., K. Macdonald, S. W. Chan, J. P. Luzio, R. Simari, and P. Weissberg. 1998. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 282:290-293. - PubMed
-
- Caelles, C., A. Helmberg, and M. Karin. 1994. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370:220-223. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous