

Progress and problems in the biology, diagnostics, and therapeutics of prion diseases
- PMID: 15254579
- PMCID: PMC449758
- DOI: 10.1172/JCI22438
Progress and problems in the biology, diagnostics, and therapeutics of prion diseases
Abstract
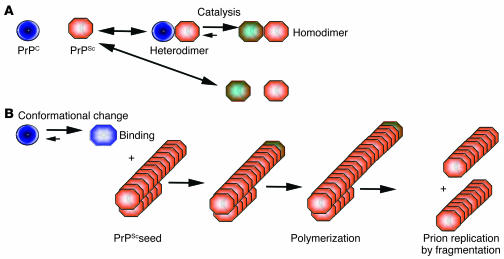
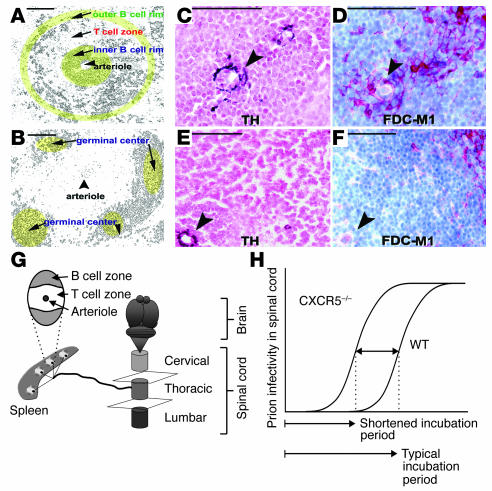
The term "prion" was introduced by Stanley Prusiner in 1982 to describe the atypical infectious agent that causes transmissible spongiform encephalopathies, a group of infectious neurodegenerative diseases that include scrapie in sheep, Creutzfeldt-Jakob disease in humans, chronic wasting disease in cervids, and bovine spongiform encephalopathy in cattle. Over the past twenty years, the word "prion" has been taken to signify various subtly different concepts. In this article, we refer to the prion as the transmissible principle underlying prion diseases, without necessarily implying any specific biochemical or structural identity. When Prusiner started his seminal work, the study of transmissible spongiform encephalopathies was undertaken by only a handful of scientists. Since that time, the "mad cow" crisis has put prion diseases on the agenda of both politicians and the media. Significant progress has been made in prion disease research, and many aspects of prion pathogenesis are now understood. And yet the diagnostic procedures available for prion diseases are not nearly as sensitive as they ought to be, and no therapeutic intervention has been shown to reliably affect the course of the diseases. This article reviews recent progress in the areas of pathogenesis of, diagnostics of, and therapy for prion diseases and highlights some conspicuous problems that remain to be addressed in each of these fields.
Figures
References
-
- Glatzel M, et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurol. 2003;2:757–763. - PubMed
-
- Collins PS, Lawson VA, Masters PC. Transmissible spongiform encephalopathies. Lancet. 2004;363:51–61. - PubMed
-
- Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. - PubMed
-
- Safar JG, et al. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat. Biotechnol. 2002;20:1147–1150. - PubMed
-
- Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Zeitschrift für die gesamte Neurologie und Psychiatrie. 1920;57:1–19. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
