

Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome
- PMID: 15254584
- PMCID: PMC449744
- DOI: 10.1172/JCI20641
Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome
Abstract
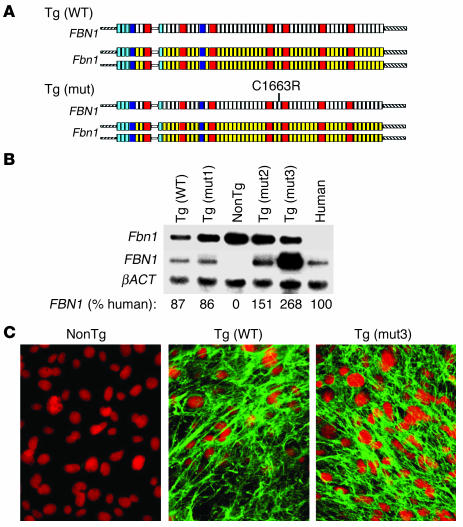
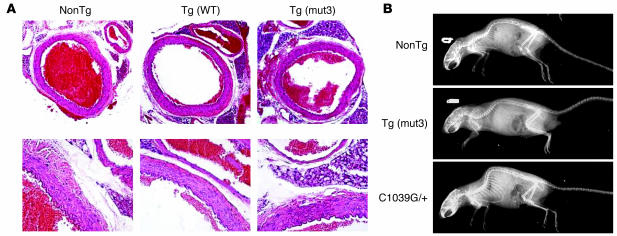
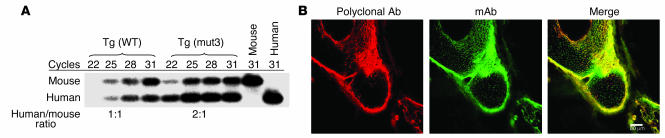
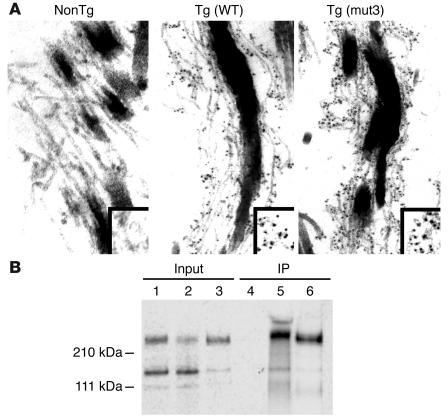
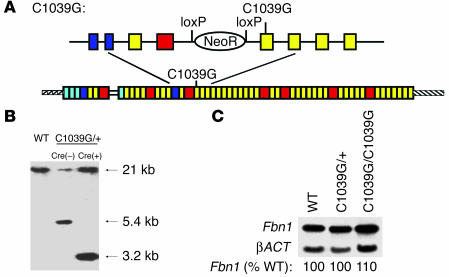
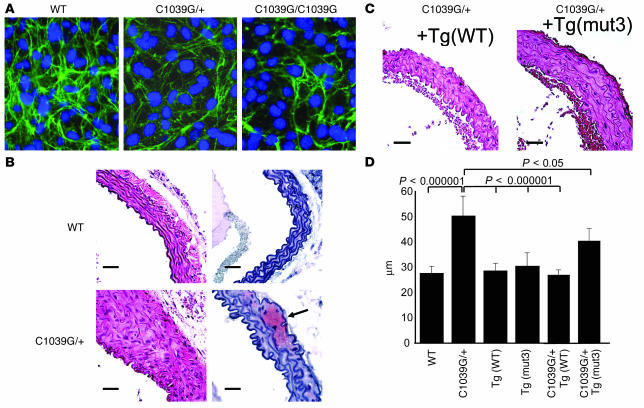
Marfan syndrome is a connective tissue disorder caused by mutations in the gene encoding fibrillin-1 (FBN1). A dominant-negative mechanism has been inferred based upon dominant inheritance, mulitimerization of monomers to form microfibrils, and the dramatic paucity of matrix-incorporated fibrillin-1 seen in heterozygous patient samples. Yeast artificial chromosome-based transgenesis was used to overexpress a disease-associated mutant form of human fibrillin-1 (C1663R) on a normal mouse background. Remarkably, these mice failed to show any abnormalities of cellular or clinical phenotype despite regulated overexpression of mutant protein in relevant tissues and developmental stages and direct evidence that mouse and human fibrillin-1 interact with high efficiency. Immunostaining with a human-specific mAb provides what we believe to be the first demonstration that mutant fibrillin-1 can participate in productive microfibrillar assembly. Informatively, use of homologous recombination to generate mice heterozygous for a comparable missense mutation (C1039G) revealed impaired microfibrillar deposition, skeletal deformity, and progressive deterioration of aortic wall architecture, comparable to characteristics of the human condition. These data are consistent with a model that invokes haploinsufficiency for WT fibrillin-1, rather than production of mutant protein, as the primary determinant of failed microfibrillar assembly. In keeping with this model, introduction of a WT FBN1 transgene on a heterozygous C1039G background rescues aortic phenotype.
Figures
Comment in
-
Determination of the molecular basis of Marfan syndrome: a growth industry.J Clin Invest. 2004 Jul;114(2):161-3. doi: 10.1172/JCI22399. J Clin Invest. 2004. PMID: 15254580 Free PMC article.
References
-
- Pyeritz, R.E. 1997. Marfan syndrome and other disorders of fibrillin. In Principles and practice of medical genetics. D.L. Rimoin, J.M. Connor, and R.E. Pyeritz, editors. Churchill Livingstone. New York, New York, USA
-
- Silverman DI, et al. Life expectancy in the Marfan syndrome. Am. J. Cardiol. 1995;75:157–160. - PubMed
-
- Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. - PubMed
-
- Dietz HC, et al. The question of heterogeneity in Marfan syndrome. Nat. Genet. 1995;9:228–231. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
