

Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration
- PMID: 15258290
- PMCID: PMC503752
- DOI: 10.1073/pnas.0403925101
Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration
Erratum in
- Proc Natl Acad Sci U S A. 2004 Oct 19;101(42):15271
Abstract
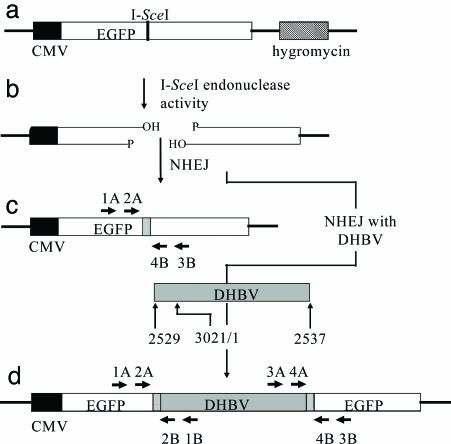
Integrated hepadnaviral DNA in livers and tumors of chronic hepatitis B patients has been reported for many years. In this study, we investigated whether hepatitis B virus DNA integration occurs preferentially at sites of cell DNA damage. A single I-SceI homing endonuclease recognition site was introduced into the DNA of the chicken hepatoma cell line LMH by stable DNA transfection, and double-strand breaks were induced by transient expression of I-SceI after transfection of an I-SceI expression vector. Alteration of the target cleavage site by imprecise nonhomologous end joining occurred at a frequency of approximately 10(-3) per transfected cell. When replication of an avian hepadnavirus, duck hepatitis B virus, occurred at the time of double-strand break repair, we observed integration of viral DNA at the site of the break with a frequency of approximately 10(-4) per transfected cell. Integration depended on the production of viral double-stranded linear DNA and the expression of I-SceI, and integrated DNA was stable through at least 17 cell divisions. Integration appeared to occur through nonhomologous end joining between the viral linear DNA ends and the I-SceI-induced break, because small deletions or insertions were observed at the sites of end joining. The results suggest that integration of hepadnaviral DNA in infected livers occurs at sites of DNA damage and may indicate the presence of more widespread genetic changes beyond that caused by viral DNA integration itself [corrected].
Figures



References
-
- Beasley, R. P. (1988) Cancer 61, 1942–1956. - PubMed
-
- Levy, L., Renard, C. A., Wei, Y. & Buendia, M. A. (2002) Ann. N.Y. Acad. Sci. 963, 21–36. - PubMed
-
- Birrer, R. B., Birrer, D. & Klavins, J. V. (2003) Ann. Clin. Lab. Sci. 33, 39–54. - PubMed
-
- Summers, J. & Mason, W. S. (1982) Cell 29, 403–415. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
