

SH2D1A regulates T-dependent humoral autoimmunity
- PMID: 15263031
- PMCID: PMC2212015
- DOI: 10.1084/jem.20040526
SH2D1A regulates T-dependent humoral autoimmunity
Abstract
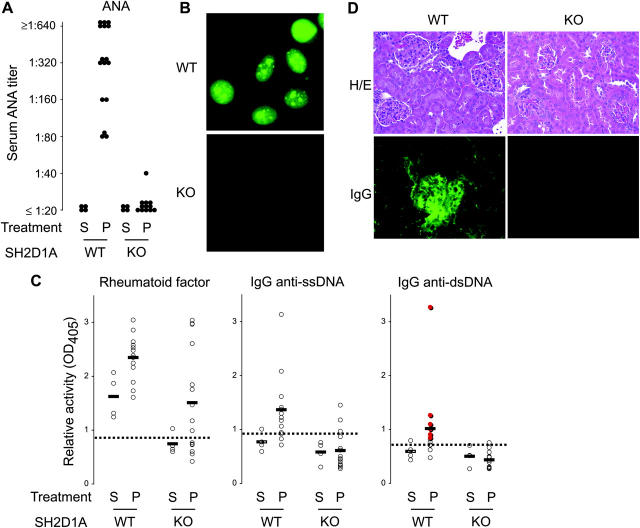
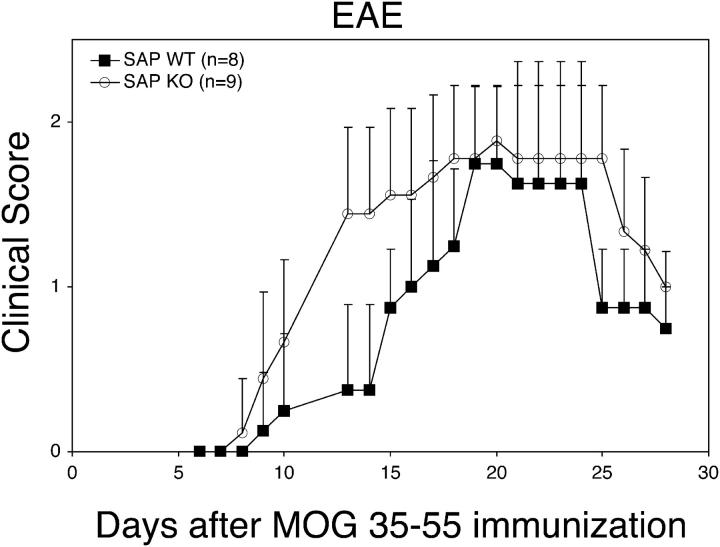
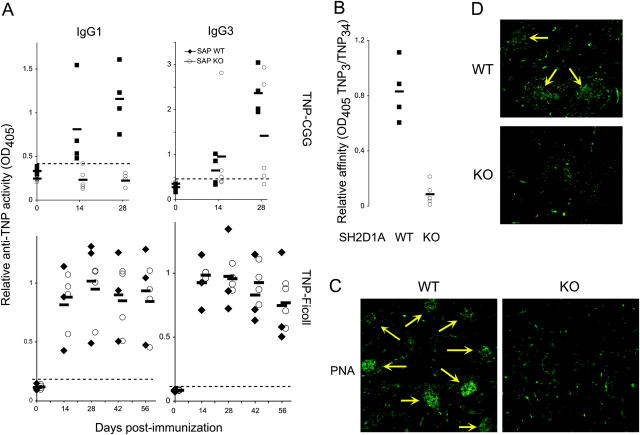
The signaling lymphocytic activation molecule (SLAM)/CD150 family includes a family of chromosome 1-encoded cell surface molecules with costimulatory functions mediated in part by the adaptor protein SH2D1A (SLAM-associated protein, SAP). Deficiency in SH2D1A protects mice from an experimental model of lupus, including the development of hypergammaglobulinemia, autoantibodies including anti-double stranded DNA, and renal disease. This protection did not reflect grossly defective T or B cell function per se because SH2D1A-deficient mice were susceptible to experimental autoimmune encephalomyelitis, a T cell-dependent disease, and they were capable of mounting normal T-independent antigen-specific immunoglobulin responses. Instead, T-dependent antibody responses were impaired in SH2D1A-deficient mice, reflecting defective germinal center formation. These findings demonstrate a specific role for the SLAM-SH2D1A system in the regulation of T-dependent humoral immune responses, implicating members of the CD150-SH2D1A family as targets in the pathogenesis and therapy of antibody-mediated autoimmune and allergic diseases.
Figures
References
-
- Engel, P., M.J. Eck, and C. Terhorst. 2003. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 3:813–821. - PubMed
-
- Cocks, B.G., C.C. Chang, J.M. Carballido, H. Yssel, J.E. de Vries, and G. Aversa. 1995. A novel receptor involved in T-cell activation. Nature. 376:260–263. - PubMed
-
- Czar, M.J., E.N. Kersh, L.A. Mijares, G. Lanier, J. Lewis, G. Yap, A. Chen, A. Sher, C.S. Duckett, R. Ahmed, et al. 2001. Altered lymphocyte responses and cytokine production in mice deficient in the X-linked lymphoproliferative disease gene SH2D1A/DSHP/SAP. Proc. Natl. Acad. Sci. USA. 98:7449–7454. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
