

State-dependent trapping of flecainide in the cardiac sodium channel
- PMID: 15272045
- PMCID: PMC1665201
- DOI: 10.1113/jphysiol.2004.065003
State-dependent trapping of flecainide in the cardiac sodium channel
Abstract
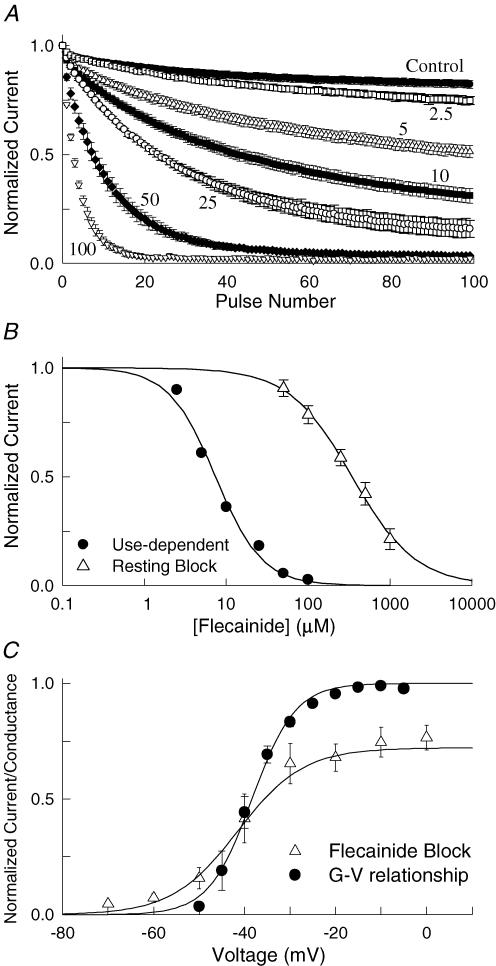
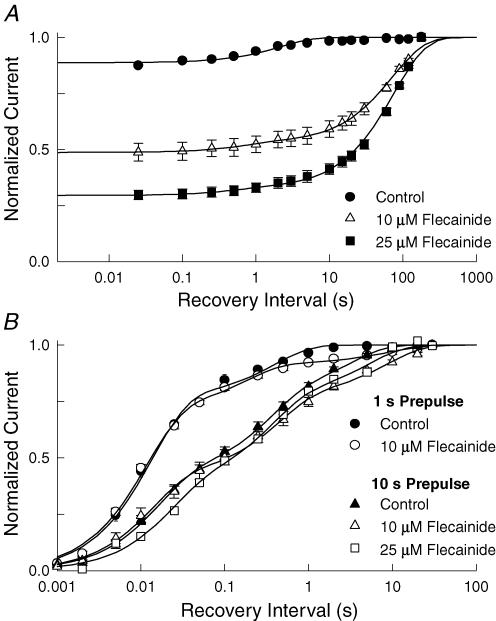
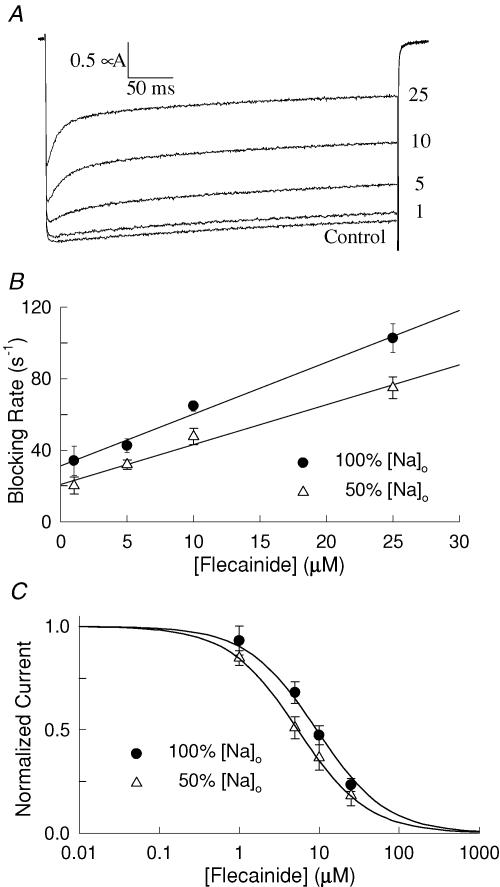
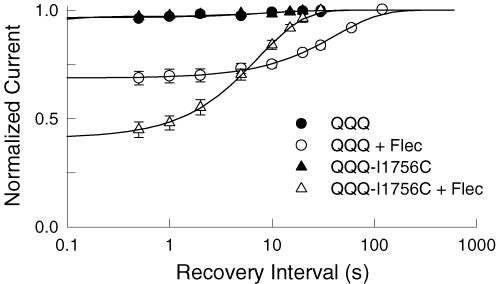
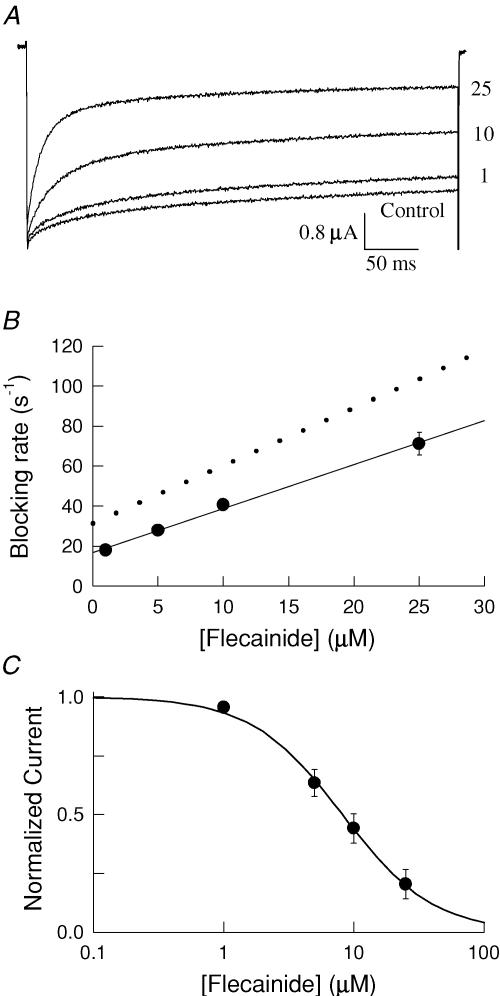
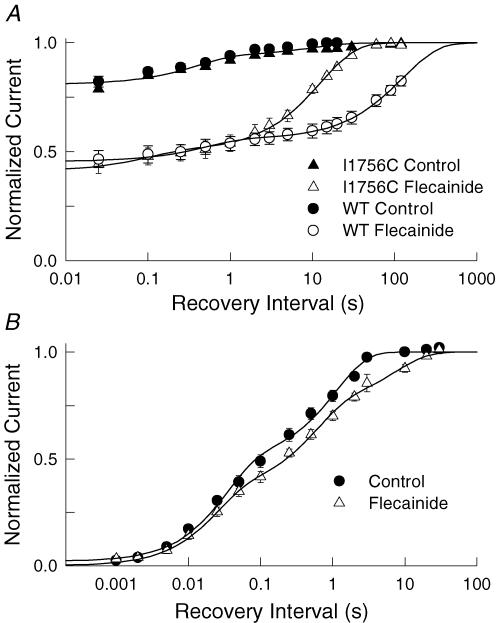
Flecainide is a Class I antiarrhythmic drug and a potent inhibitor of the cardiac (Nav1.5) sodium channel. Although the flecainide inhibition of Nav1.5 is typically enhanced by depolarization, the contributions of the open and inactivated states to flecainide binding and inhibition remain controversial. We further investigated the state-dependent binding of flecainide by examining its inhibition of rapidly inactivating and non-inactivating mutants of Nav1.5 expressed in Xenopus oocytes. Applying flecainide while briefly depolarizing from a relatively negative holding potential resulted in a low-affinity inhibition of the channel (IC(50) = 345 microM). Increasing the frequency of stimulation potentiated the flecainide inhibition (IC(50) = 7.4 microM), which progressively increased over the range of voltages where Nav1.5 channels activated. This contrasts with sustained depolarizations that effectively stabilize the channels in inactivated states, which failed to promote significant flecainide inhibition. The voltage sensitivity and strong dependence of the flecainide inhibition on repetitive depolarization suggests that flecainide binding is facilitated by channel opening and that the drug does not directly bind to closed or inactivated channels. The binding of flecainide to open channels was further investigated in a non-inactivating mutant of Nav1.5. Flecainide produced a time-dependent decay in the current of the non-inactivating mutant that displayed kinetics consistent with a simple pore blocking mechanism (K(D) = 11 microM). At hyperpolarized voltages, flecainide slowed the recovery of both the rapidly inactivating (tau = 81 +/- 3 s) and non-inactivating (tau = 42 +/- 3 s) channels. Mutation of a conserved isoleucine of the D4S6 segment (I1756C) creates an alternative pathway that permits the rapid diffusion of anaesthetics out of the Nav1.5 channel. The I1756C mutation accelerated the recovery of both the rapidly inactivating (tau = 12.6 +/- 0.4 s) and non-inactivating (tau = 7.4 +/- 0.1 s) channels, suggesting that flecainide is trapped and not tightly bound within the pore when the channels are closed or inactivated. The data indicate that flecainide rapidly gains access to its binding site when the channel is open and inhibits Na(+) current by a pore blocking mechanism. Closing of either the activation or the inactivation gate traps flecainide within the pore resulting in the slow recovery of the drug-modified channels at hyperpolarized voltages.
Figures
Similar articles
-
Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels.J Physiol. 2002 Jun 15;541(Pt 3):701-16. doi: 10.1113/jphysiol.2001.016139. J Physiol. 2002. PMID: 12068034 Free PMC article.
-
Class Ic antiarrhythmics block human skeletal muscle Na channel during myotonia-like stimulation.Eur J Pharmacol. 2006 Feb 17;532(1-2):24-31. doi: 10.1016/j.ejphar.2005.12.021. Epub 2006 Feb 10. Eur J Pharmacol. 2006. PMID: 16473348
-
Closing and inactivation potentiate the cocaethylene inhibition of cardiac sodium channels by distinct mechanisms.Mol Pharmacol. 2003 Dec;64(6):1575-85. doi: 10.1124/mol.64.6.1575. Mol Pharmacol. 2003. PMID: 14645689
-
Mutations in cardiac sodium channels: clinical implications.Am J Pharmacogenomics. 2003;3(3):173-9. doi: 10.2165/00129785-200303030-00003. Am J Pharmacogenomics. 2003. PMID: 12814325 Review.
-
Molecular mechanisms of local anesthesia: a review.Anesthesiology. 1990 Apr;72(4):711-34. doi: 10.1097/00000542-199004000-00022. Anesthesiology. 1990. PMID: 2157353 Review.
Cited by
-
Flecainide Toxicity Resulting in Pacemaker Latency and Intermittent Failure to Capture.Am J Case Rep. 2019 Aug 30;20:1279-1283. doi: 10.12659/AJCR.916370. Am J Case Rep. 2019. PMID: 31467262 Free PMC article.
-
Pharmacology of A-Type K+ Channels.Handb Exp Pharmacol. 2021;267:167-183. doi: 10.1007/164_2021_456. Handb Exp Pharmacol. 2021. PMID: 33907894
-
The Antiarrhythmic Mechanisms of Flecainide in Catecholaminergic Polymorphic Ventricular Tachycardia.Front Physiol. 2022 Mar 9;13:850117. doi: 10.3389/fphys.2022.850117. eCollection 2022. Front Physiol. 2022. PMID: 35356081 Free PMC article. Review.
-
Dynamics of sodium channel Nav1.5 expression in astrocytes in mouse models of multiple sclerosis.Neuroreport. 2014 Oct 22;25(15):1208-15. doi: 10.1097/WNR.0000000000000249. Neuroreport. 2014. PMID: 25144393 Free PMC article.
-
Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/- murine hearts modelling the Brugada syndrome.J Physiol. 2007 May 15;581(Pt 1):255-75. doi: 10.1113/jphysiol.2007.128785. Epub 2007 Feb 15. J Physiol. 2007. PMID: 17303635 Free PMC article.
References
-
- Anderson J, Gilbert E, Alpert B, Henthorn R, Waldo A, Bhandari A, Hawkinson R, Pritchett E. Prevention of symptomatic recurrences of paroxysmal atrial fibrillation in patients initially tolerating antiarrhythmic therapy. A multicenter, double-blind, crossover study of flecainide and placebo with transtelephonic monitoring. Flecainide Supraventricular Tachycardia Study Group. Circ. 1989;80:557–1570. - PubMed
-
- Anno T, Hondeghem L. Interactions of flecainide with guinea pig cardiac sodium channels. Importance of activation unblocking to the voltage dependence of recovery. Circ Res. 1990;66:89–803. - PubMed
-
- Benhorin J, Taub R, Goldmit M, Kerem B, Kass R, Windman I, Medina A. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long–QT syndrome? Circ. 2000;101:1698–1706. - PubMed
-
- Brugada R, Brugada J, Antzelevitch C, Kirsch G, Potenza D, Towbin J, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circ. 2000;101:510–515. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous