

Graph-based iterative Group Analysis enhances microarray interpretation
- PMID: 15272936
- PMCID: PMC509016
- DOI: 10.1186/1471-2105-5-100
Graph-based iterative Group Analysis enhances microarray interpretation
Abstract
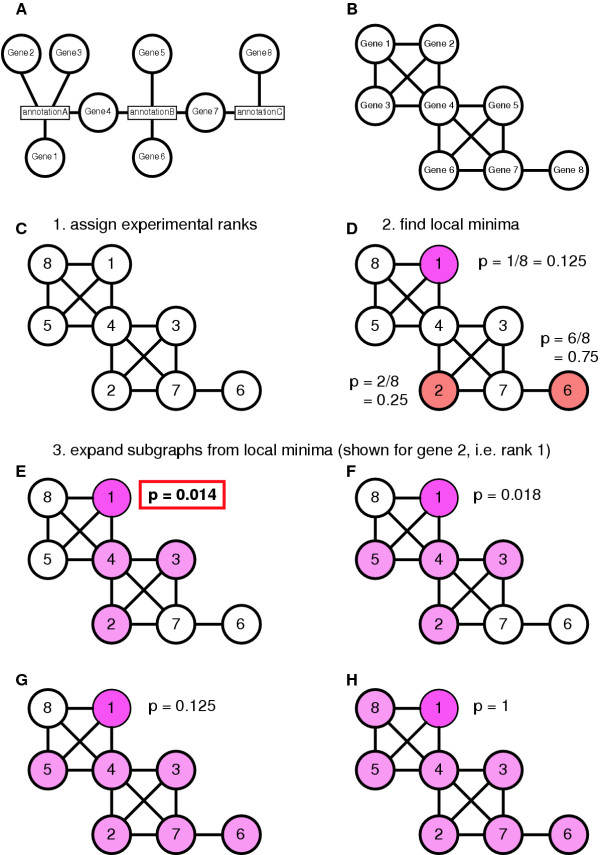
Background: One of the most time-consuming tasks after performing a gene expression experiment is the biological interpretation of the results by identifying physiologically important associations between the differentially expressed genes. A large part of the relevant functional evidence can be represented in the form of graphs, e.g. metabolic and signaling pathways, protein interaction maps, shared GeneOntology annotations, or literature co-citation relations. Such graphs are easily constructed from available genome annotation data. The problem of biological interpretation can then be described as identifying the subgraphs showing the most significant patterns of gene expression. We applied a graph-based extension of our iterative Group Analysis (iGA) approach to obtain a statistically rigorous identification of the subgraphs of interest in any evidence graph.
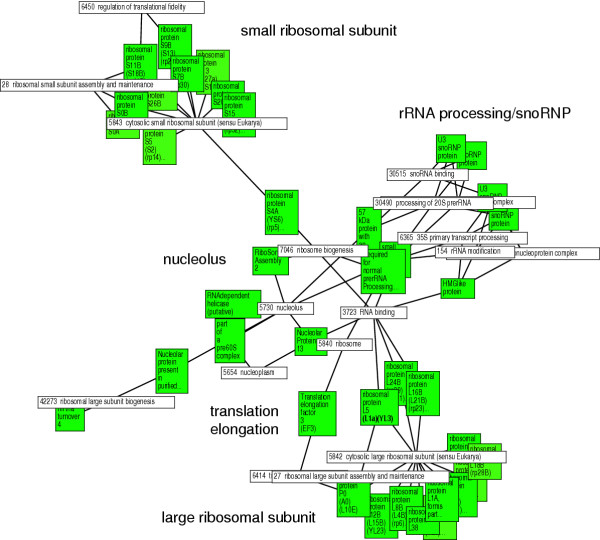
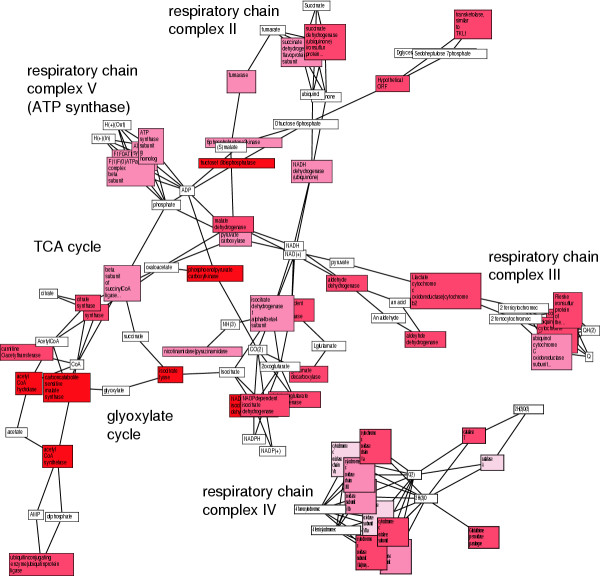
Results: We validated the Graph-based iterative Group Analysis (GiGA) by applying it to the classic yeast diauxic shift experiment of DeRisi et al., using GeneOntology and metabolic network information. GiGA reliably identified and summarized all the biological processes discussed in the original publication. Visualization of the detected subgraphs allowed the convenient exploration of the results. The method also identified several processes that were not presented in the original paper but are of obvious relevance to the yeast starvation response.
Conclusions: GiGA provides a fast and flexible delimitation of the most interesting areas in a microarray experiment, and leads to a considerable speed-up and improvement of the interpretation process.
Figures
References
-
- Dudoit S, Yang YH, Callow MJ, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica. 2002;12:111–139.
-
- Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Letters. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
