

G protein-coupled receptors: in silico drug discovery in 3D
- PMID: 15277683
- PMCID: PMC509175
- DOI: 10.1073/pnas.0401862101
G protein-coupled receptors: in silico drug discovery in 3D
Abstract
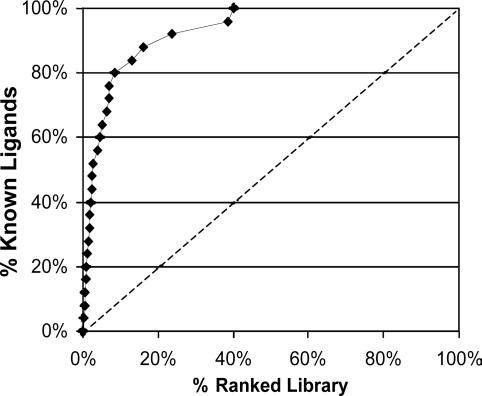
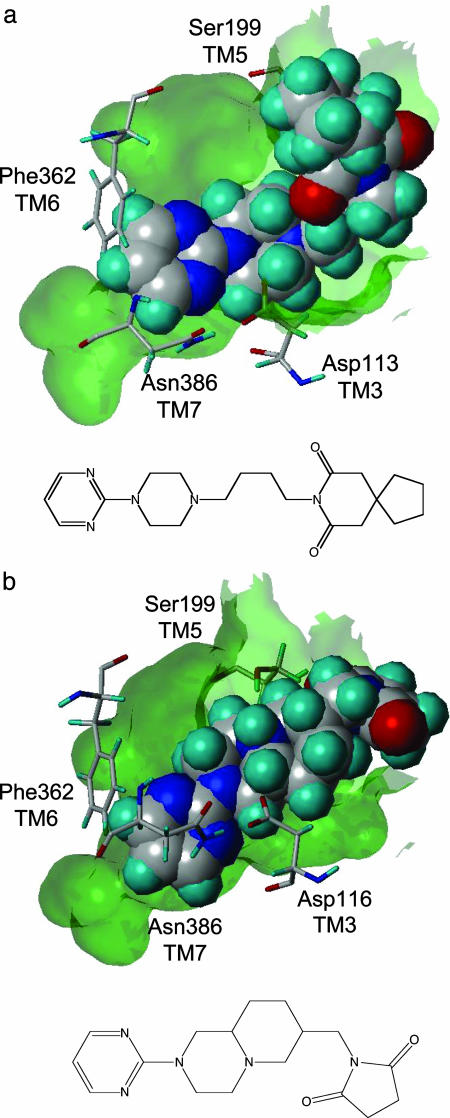
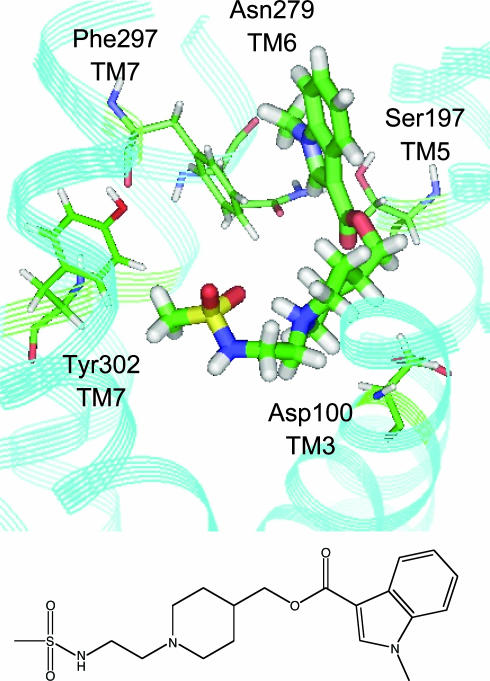
The application of structure-based in silico methods to drug discovery is still considered a major challenge, especially when the x-ray structure of the target protein is unknown. Such is the case with human G protein-coupled receptors (GPCRs), one of the most important families of drug targets, where in the absence of x-ray structures, one has to rely on in silico 3D models. We report repeated success in using ab initio in silico GPCR models, generated by the predict method, for blind in silico screening when applied to a set of five different GPCR drug targets. More than 100,000 compounds were typically screened in silico for each target, leading to a selection of <100 "virtual hit" compounds to be tested in the lab. In vitro binding assays of the selected compounds confirm high hit rates, of 12-21% (full dose-response curves, Ki < 5 microM). In most cases, the best hit was a novel compound (New Chemical Entity) in the 1- to 100-nM range, with very promising pharmacological properties, as measured by a variety of in vitro and in vivo assays. These assays validated the quality of the hits as lead compounds for drug discovery. The results demonstrate the usefulness and robustness of ab initio in silico 3D models and of in silico screening for GPCR drug discovery.
Figures
References
-
- Wilson, S. & Bergsma, D. (2000) Drug Des. Discovery 17, 105–114. - PubMed
-
- Sautel, M. & Milligan, G. (2000) Curr. Med. Chem. 7, 889–896. - PubMed
-
- Schoneberg, T., Schulz, A. & Gudermann, T. (2002) Rev. Physiol. Biochem. Pharmacol. 144, 143–227. - PubMed
-
- Rattner, A., Sun, H. & Nathans, J. (1999) Annu. Rev. Genet. 33, 89–131. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
