

Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus
- PMID: 15280481
- PMCID: PMC479062
- DOI: 10.1128/JVI.78.16.8732-8745.2004
Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus
Abstract
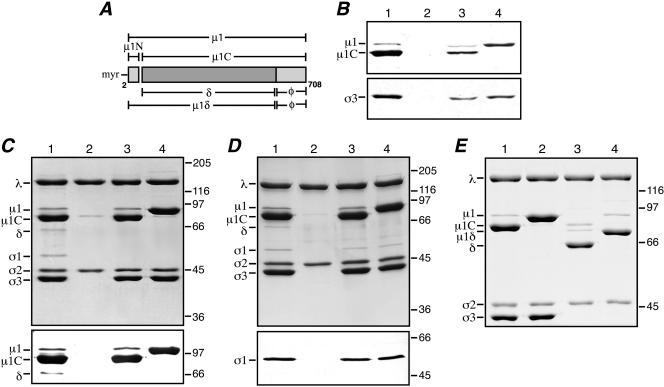
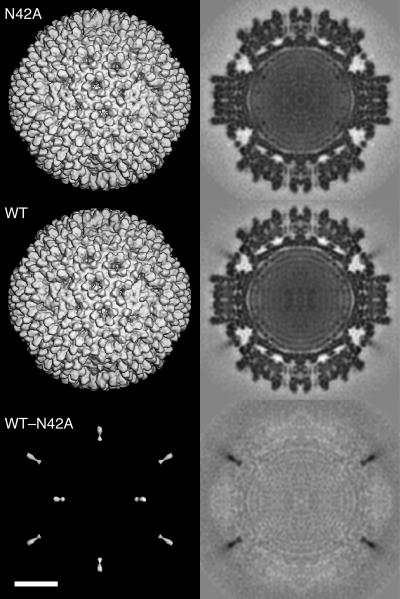
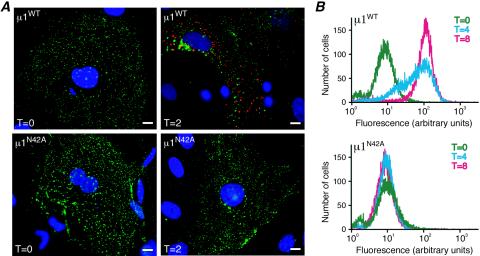
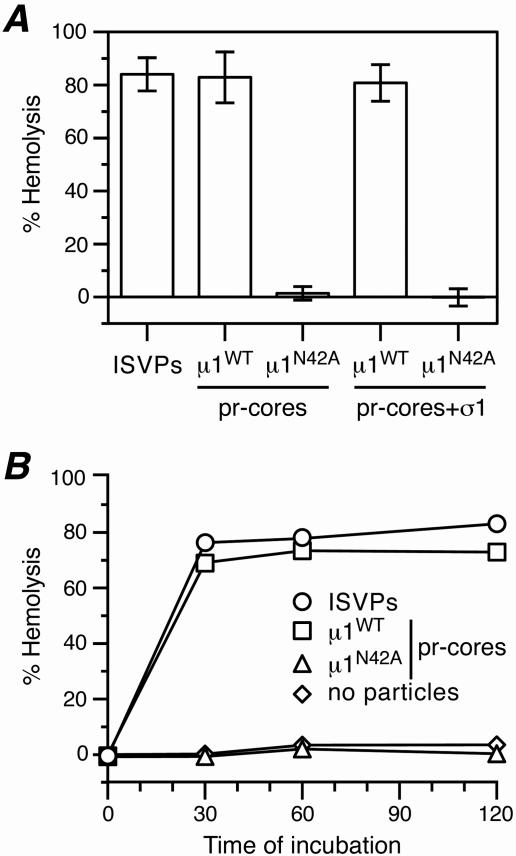
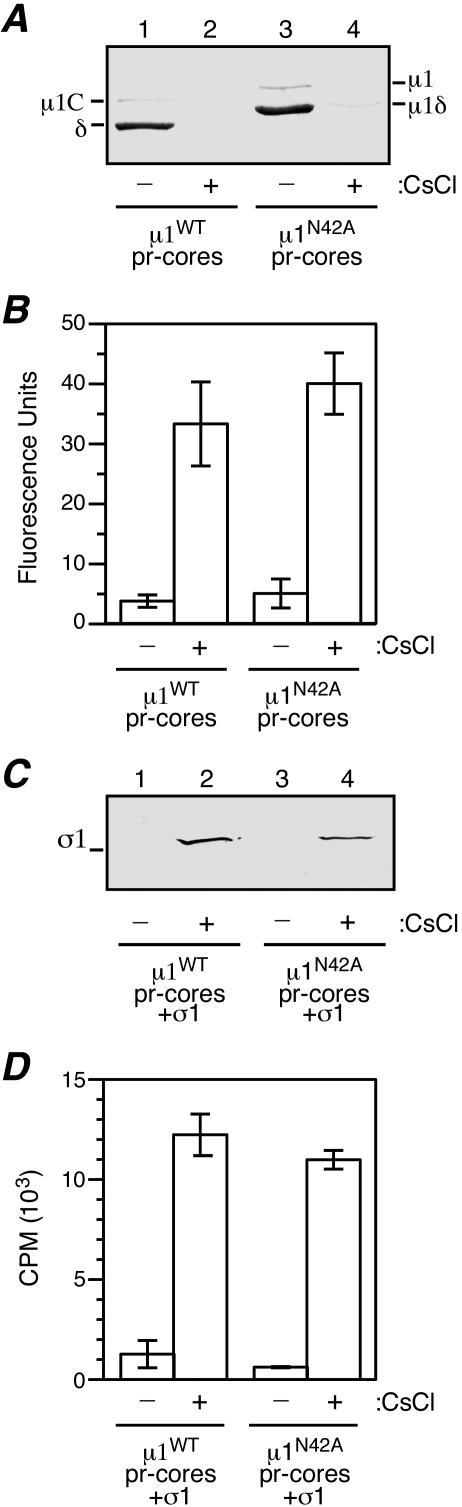
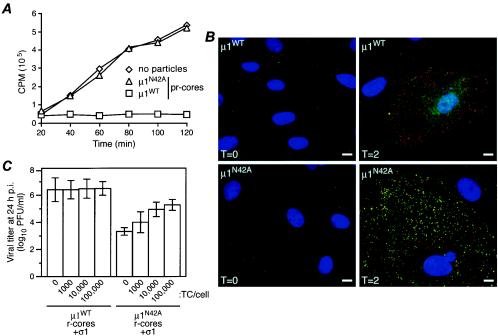
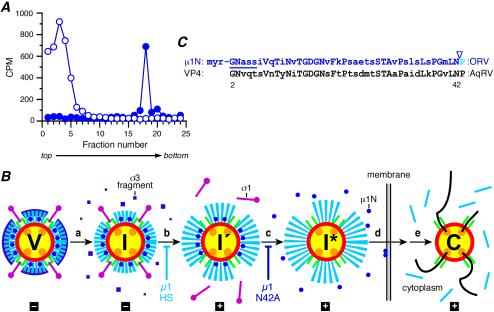
Several nonenveloped animal viruses possess an autolytic capsid protein that is cleaved as a maturation step during assembly to yield infectious virions. The 76-kDa major outer capsid protein micro1 of mammalian orthoreoviruses (reoviruses) is also thought to be autocatalytically cleaved, yielding the virion-associated fragments micro1N (4 kDa; myristoylated) and micro1C (72 kDa). In this study, we found that micro1 cleavage to yield micro1N and micro1C was not required for outer capsid assembly but contributed greatly to the infectivity of the assembled particles. Recoated particles containing mutant, cleavage-defective micro1 (asparagine --> alanine substitution at amino acid 42) were competent for attachment; processing by exogenous proteases; structural changes in the outer capsid, including micro1 conformational change and sigma1 release; and transcriptase activation but failed to mediate membrane permeabilization either in vitro (no hemolysis) or in vivo (no coentry of the ribonucleotoxin alpha-sarcin). In addition, after these particles were allowed to enter cells, the delta region of micro1 continued to colocalize with viral core proteins in punctate structures, indicating that both elements remained bound together in particles and/or trapped within the same subcellular compartments, consistent with a defect in membrane penetration. If membrane penetration activity was supplied in trans by a coinfecting genome-deficient particle, the recoated particles with cleavage-defective micro1 displayed much higher levels of infectivity. These findings led us to propose a new uncoating intermediate, at which particles are trapped in the absence of micro1N/micro1C cleavage. We additionally showed that this cleavage allowed the myristoylated, N-terminal micro1N fragment to be released from reovirus particles during entry-related uncoating, analogous to the myristoylated, N-terminal VP4 fragment of picornavirus capsid proteins. The results thus suggest that hydrophobic peptide release following capsid protein autocleavage is part of a general mechanism of membrane penetration shared by several diverse nonenveloped animal viruses.
Figures
References
-
- Attoui, H., Q. Fang, F. M. Jaafar, J. F. Cantaloube, P. Biagini, P. De Micco, and X. De Lamballerie. 2002. Common evolutionary origin of aquareoviruses and orthoreoviruses revealed by genome characterization of Golden shiner reovirus, Grass carp reovirus, Striped bass reovirus and golden ide reovirus (genus Aquareovirus, family Reoviridae). J. Gen. Virol. 83:1941-1951. - PubMed
-
- Baker, T. S., and R. H. Cheng. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryo-electron microscopy. J. Struct. Biol. 116:120-130. - PubMed
-
- Barton, E. S., J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, A. Nusrat, C. A. Parkos, and T. S. Dermody. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441-451. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
