

Abnormal cardiac development in the absence of heart glycogen
- PMID: 15282316
- PMCID: PMC479719
- DOI: 10.1128/MCB.24.16.7179-7187.2004
Abnormal cardiac development in the absence of heart glycogen
Abstract
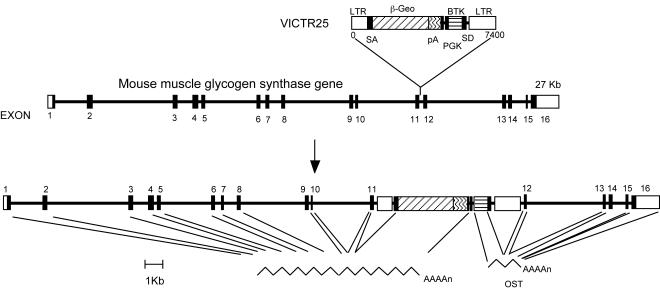
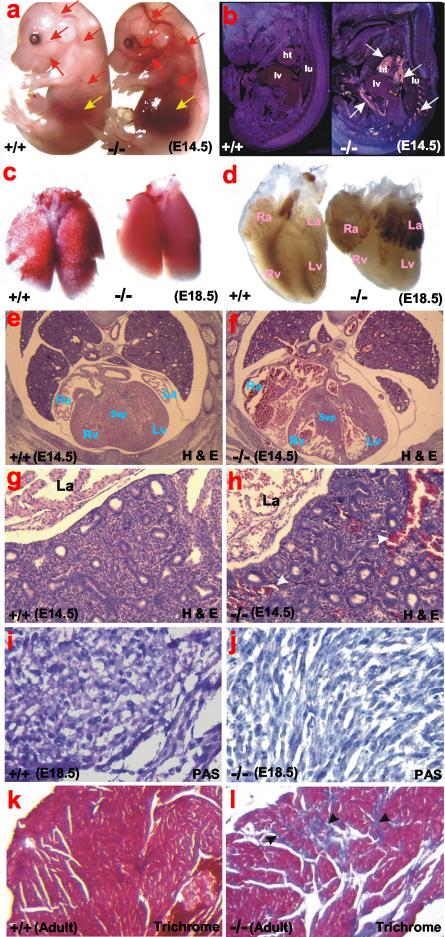
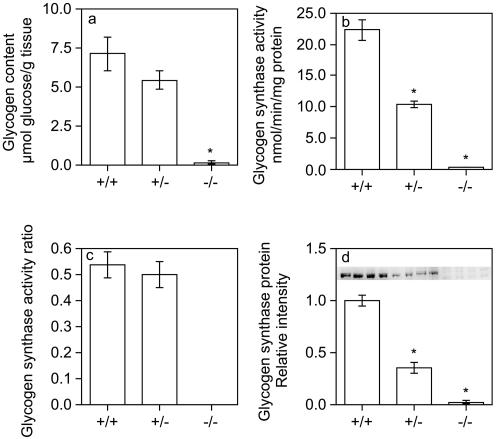
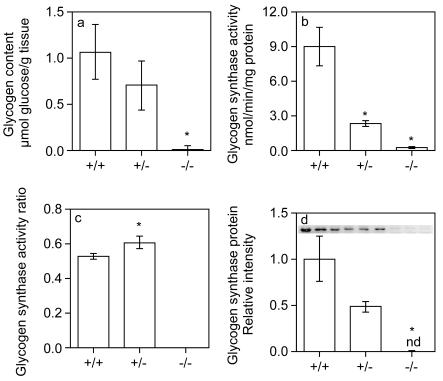
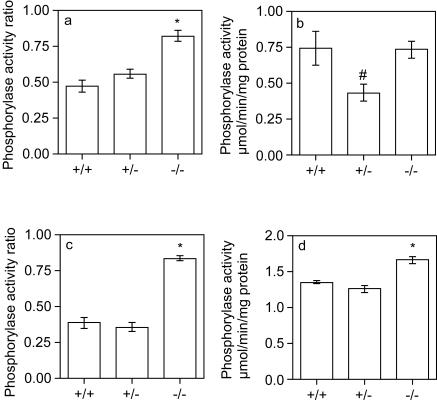
Glycogen serves as a repository of glucose in many mammalian tissues. Mice lacking this glucose reserve in muscle, heart, and several other tissues were generated by disruption of the GYS1 gene, which encodes an isoform of glycogen synthase. Crossing mice heterozygous for the GYS1 disruption resulted in a significant underrepresentation of GYS1-null mice in the offspring. Timed matings established that Mendelian inheritance was followed for up to 18.5 days postcoitum (dpc) and that approximately 90% of GYS1-null animals died soon after birth due to impaired cardiac function. Defects in cardiac development began between 11.5 and 14.5 dpc. At 18.5 dpc, the hearts were significantly smaller, with reduced ventricular chamber size and enlarged atria. Consistent with impaired cardiac function, edema, pooling of blood, and hemorrhagic liver were seen. Glycogen synthase and glycogen were undetectable in cardiac muscle and skeletal muscle from the surviving null mice, and the hearts showed normal morphology and function. Congenital heart disease is one of the most common birth defects in humans, at up to 1 in 50 live births. The results provide the first direct evidence that the ability to synthesize glycogen in cardiac muscle is critical for normal heart development and hence that its impairment could be a significant contributor to congenital heart defects.
Figures
Similar articles
-
Glycogen synthesis is required for adaptive thermogenesis in beige adipose tissue and affects diet-induced obesity.Am J Physiol Endocrinol Metab. 2024 May 1;326(5):E696-E708. doi: 10.1152/ajpendo.00074.2024. Epub 2024 Apr 3. Am J Physiol Endocrinol Metab. 2024. PMID: 38568151
-
Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease.Clin Transl Med. 2025 Apr;15(4):e70314. doi: 10.1002/ctm2.70314. Clin Transl Med. 2025. PMID: 40268518 Free PMC article.
-
Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise.J Biol Chem. 2005 Apr 29;280(17):17260-5. doi: 10.1074/jbc.M410448200. Epub 2005 Feb 11. J Biol Chem. 2005. PMID: 15711014
-
Small molecule inhibition of glycogen synthase I reduces muscle glycogen content and improves biomarkers in a mouse model of Pompe disease.Am J Physiol Endocrinol Metab. 2024 Oct 1;327(4):E524-E532. doi: 10.1152/ajpendo.00175.2024. Epub 2024 Aug 22. Am J Physiol Endocrinol Metab. 2024. PMID: 39171753
-
Studies of gene expression and activity of hexokinase, phosphofructokinase and glycogen synthase in human skeletal muscle in states of altered insulin-stimulated glucose metabolism.Dan Med Bull. 1999 Feb;46(1):13-34. Dan Med Bull. 1999. PMID: 10081651 Review.
Cited by
-
A Modified Enzymatic Method for Measurement of Glycogen Content in Glycogen Storage Disease Type IV.JIMD Rep. 2016;30:89-94. doi: 10.1007/8904_2015_522. Epub 2016 Jun 26. JIMD Rep. 2016. PMID: 27344645 Free PMC article.
-
Glycogen and its metabolism: some new developments and old themes.Biochem J. 2012 Feb 1;441(3):763-87. doi: 10.1042/BJ20111416. Biochem J. 2012. PMID: 22248338 Free PMC article. Review.
-
Impaired glucose tolerance and predisposition to the fasted state in liver glycogen synthase knock-out mice.J Biol Chem. 2010 Apr 23;285(17):12851-61. doi: 10.1074/jbc.M110.106534. Epub 2010 Feb 23. J Biol Chem. 2010. PMID: 20178984 Free PMC article.
-
A century of exercise physiology: key concepts in regulation of glycogen metabolism in skeletal muscle.Eur J Appl Physiol. 2022 Aug;122(8):1751-1772. doi: 10.1007/s00421-022-04935-1. Epub 2022 Mar 30. Eur J Appl Physiol. 2022. PMID: 35355125 Free PMC article. Review.
-
Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease.Hum Mol Genet. 2010 Feb 15;19(4):684-96. doi: 10.1093/hmg/ddp535. Epub 2009 Dec 3. Hum Mol Genet. 2010. PMID: 19959526 Free PMC article.
References
-
- Altman, P. L., and D. S. Dittmer. 1962. Growth including reproduction and morphological development. Federation of American Societies for Experimental Biology, Washington, D.C.
-
- Arad, M., I. P. Moskowitz, V. V. Patel, F. Ahmad, A. R. Perez-Atayde, D. B. Sawyer, M. Walter, G. H. Li, P. G. Burgon, C. T. Maguire, D. Stapleton, J. P. Schmitt, X. X. Guo, A. Pizard, S. Kupershmidt, D. M. Roden, C. I. Berul, C. E. Seidman, and J. G. Seidman. 2003. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107:2850-2856. - PubMed
-
- Azpiazu, I., A. R. Saltiel, A. A. DePaoli-Roach, and J. C. Lawrence. 1996. Regulation of both glycogen synthase and PHAS-I by insulin in rat skeletal muscle involves mitogen-activated protein kinase-independent and rapamycin-sensitive pathways. J. Biol. Chem. 271:5033-5039. - PubMed
-
- Bergmeyer, H. U., E. Berndt, F. Schmidt, and H. Stork. 1974. Glucose determination with hexokinase and glucose-6-phosphate dehydrogenase, p. 1196-1201. In H. U. Bergmeyer (ed.), Methods of enzymatic analysis, 2nd ed., vol. 3. Academic Press, Inc., New York, N.Y.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases