

Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis
- PMID: 15289504
- PMCID: PMC2211978
- DOI: 10.1084/jem.20040213
Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis
Abstract
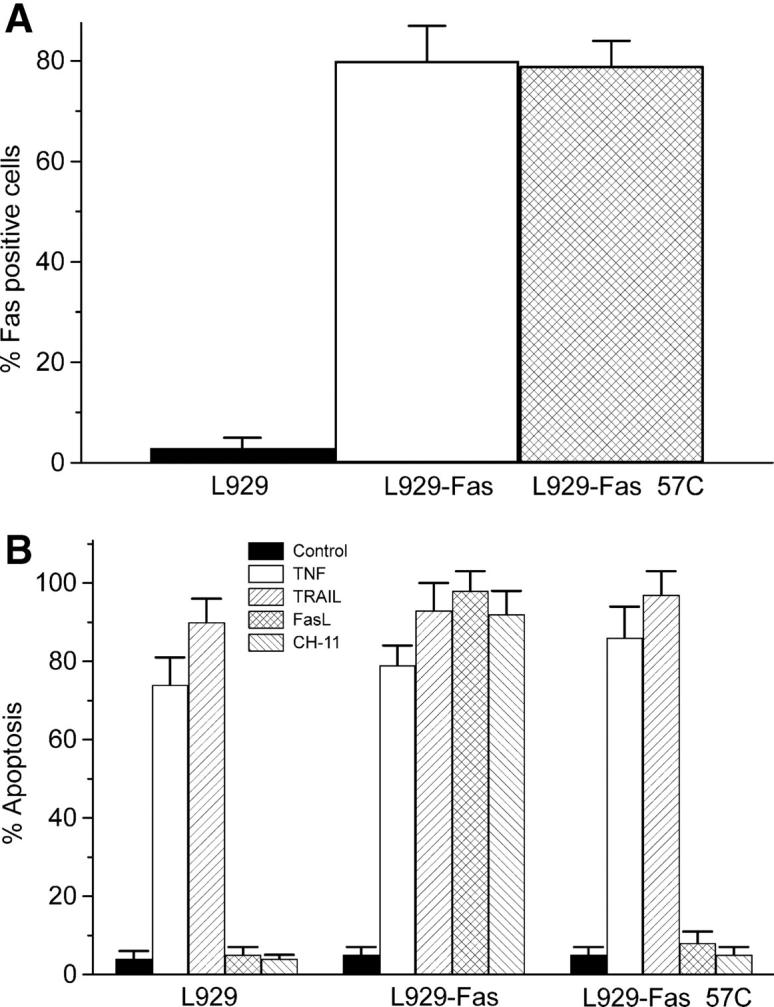
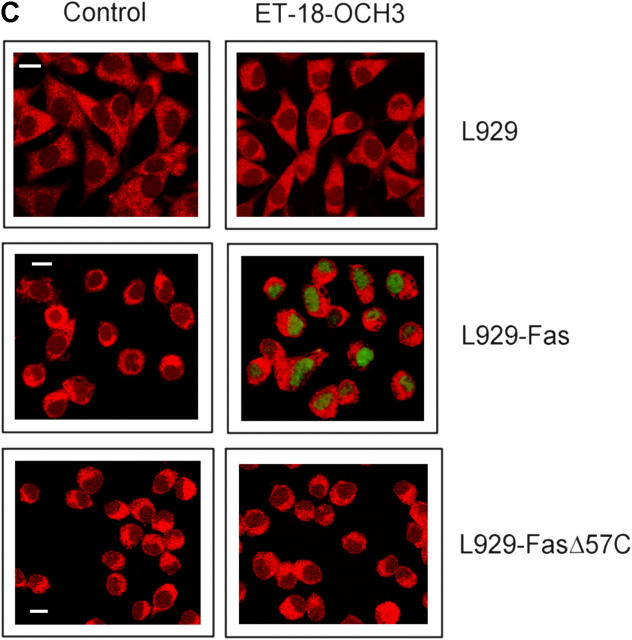
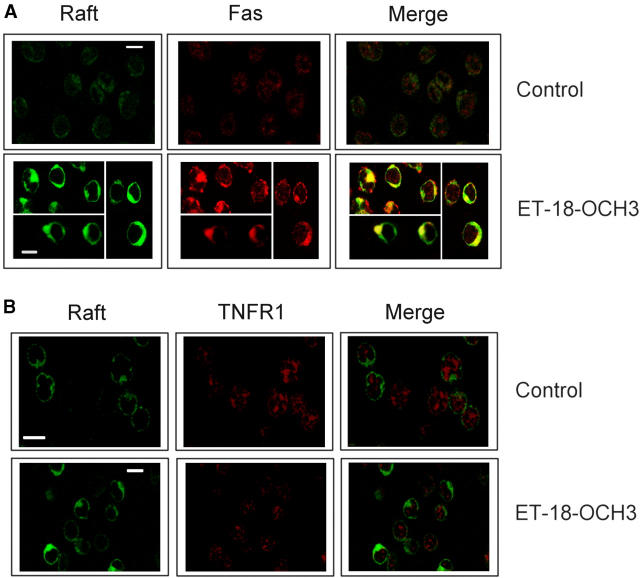
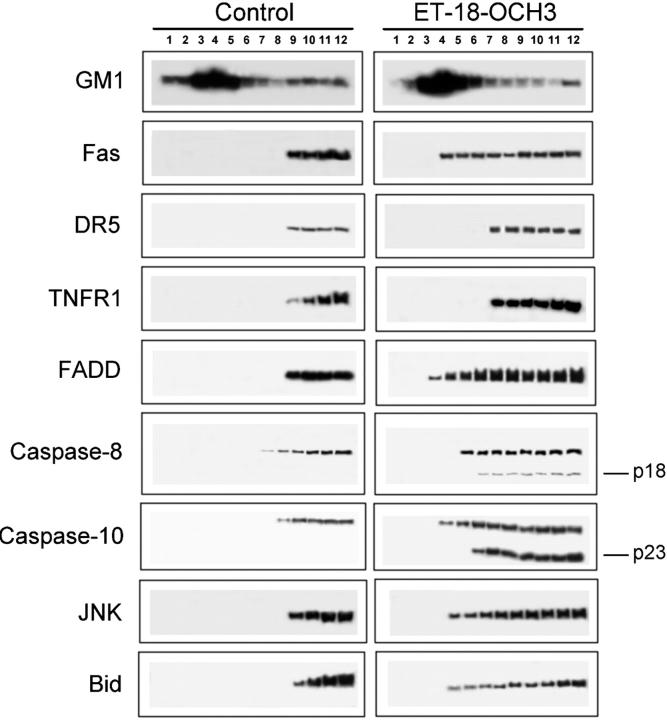
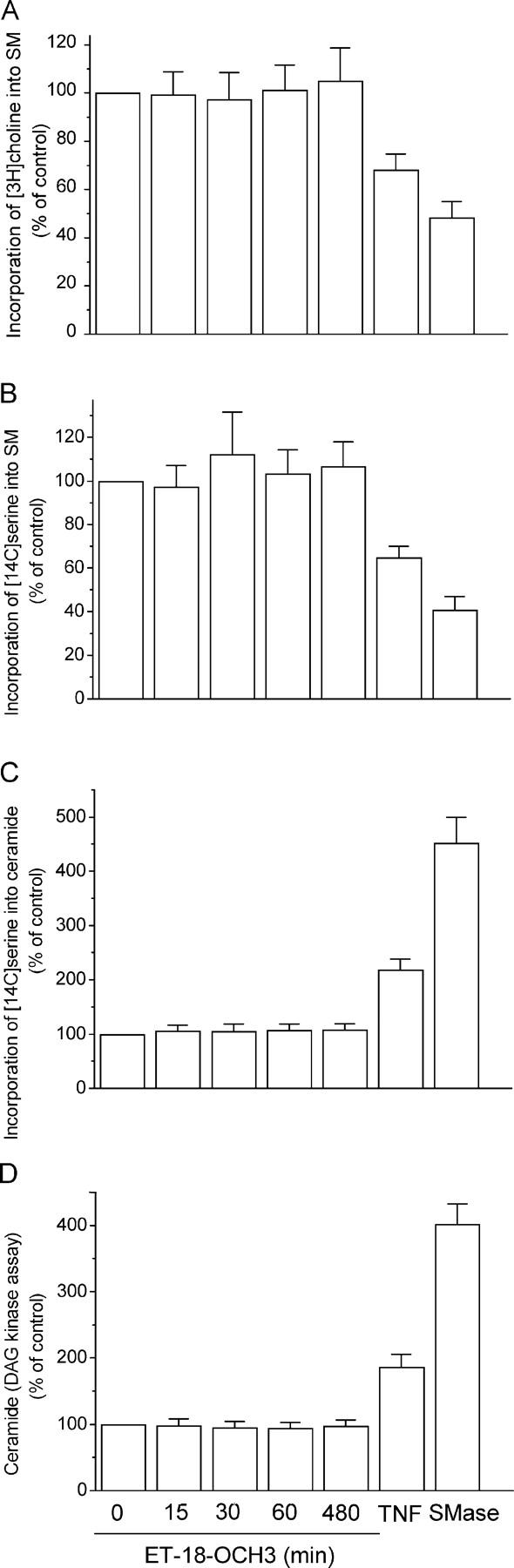
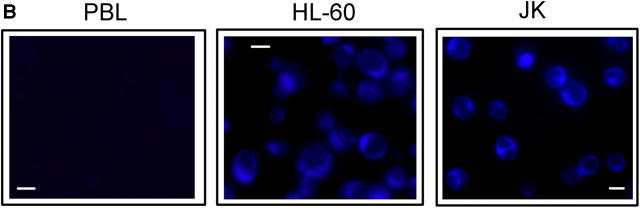
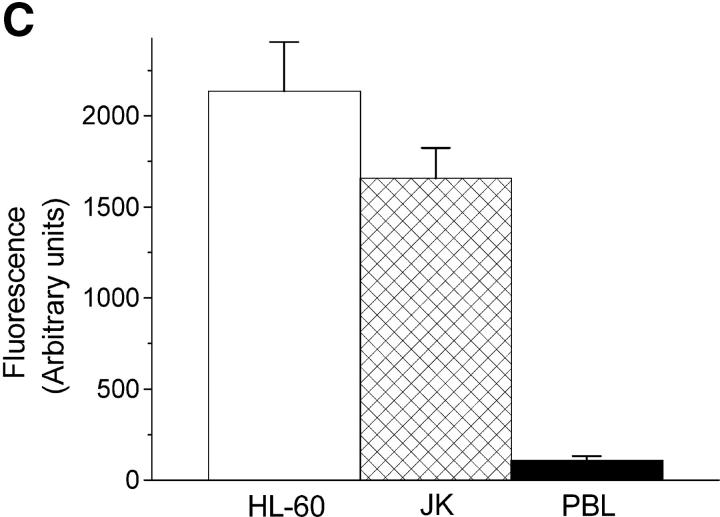
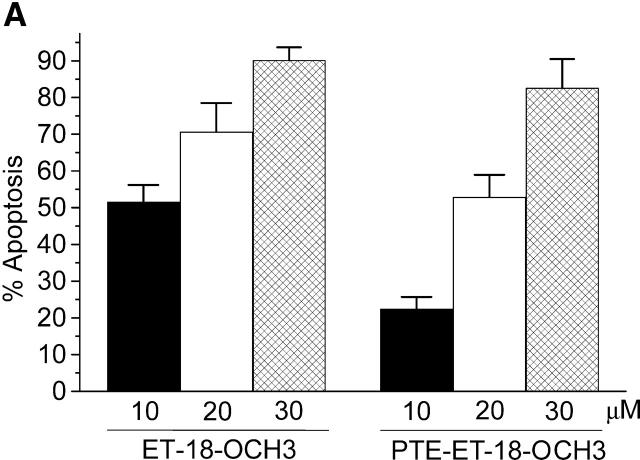
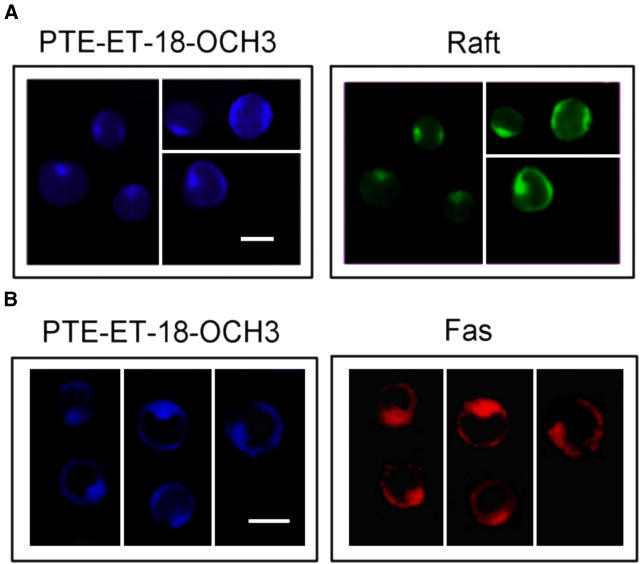
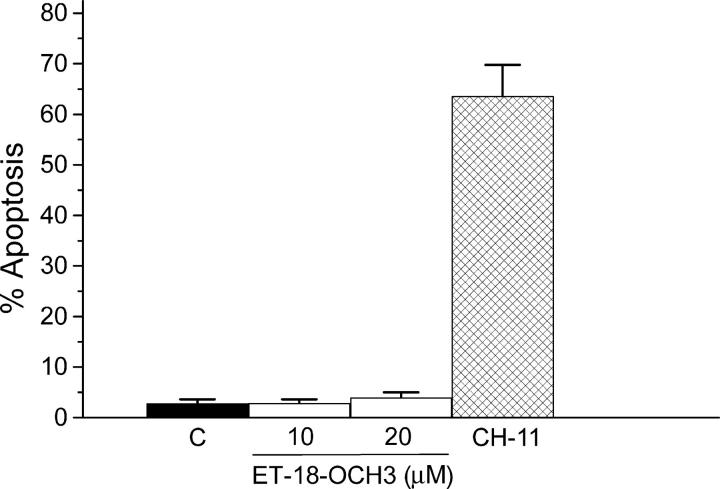
We have discovered a new and specific cell-killing mechanism mediated by the selective uptake of the antitumor drug 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (ET-18-OCH(3), Edelfosine) into lipid rafts of tumor cells, followed by its coaggregation with Fas death receptor (also known as APO-1 or CD95) and recruitment of apoptotic molecules into Fas-enriched rafts. Drug sensitivity was dependent on drug uptake and Fas expression, regardless of the presence of other major death receptors, such as tumor necrosis factor (TNF) receptor 1 or TNF-related apoptosis-inducing ligand R2/DR5 in the target cell. Drug microinjection experiments in Fas-deficient and Fas-transfected cells unable to incorporate exogenous ET-18-OCH(3) demonstrated that Fas was intracellularly activated. Partial deletion of the Fas intracellular domain prevented apoptosis. Unlike normal lymphocytes, leukemic T cells incorporated ET-18-OCH(3) into rafts coaggregating with Fas and underwent apoptosis. Fas-associated death domain protein, procaspase-8, procaspase-10, c-Jun amino-terminal kinase, and Bid were recruited into rafts, linking Fas and mitochondrial signaling routes. Clustering of rafts was necessary but not sufficient for ET-18-OCH(3)-mediated cell death, with Fas being required as the apoptosis trigger. ET-18-OCH(3)-mediated apoptosis did not require sphingomyelinase activation. Normal cells, including human and rat hepatocytes, did not incorporate ET-18-OCH(3) and were spared. This mechanism represents the first selective activation of Fas in tumor cells. Our data set a framework for the development of more targeted therapies leading to intracellular Fas activation and recruitment of downstream signaling molecules into Fas-enriched rafts.
Figures
Similar articles
-
Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy.Drug Resist Updat. 2006 Feb-Apr;9(1-2):51-73. doi: 10.1016/j.drup.2006.04.002. Epub 2006 May 9. Drug Resist Updat. 2006. PMID: 16687251 Review.
-
Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts.Blood. 2007 Jan 15;109(2):711-9. doi: 10.1182/blood-2006-04-016824. Epub 2006 Sep 26. Blood. 2007. PMID: 17003375
-
Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis.Int J Cancer. 2000 Mar 1;85(5):674-82. doi: 10.1002/(sici)1097-0215(20000301)85:5<674::aid-ijc13>3.0.co;2-z. Int J Cancer. 2000. PMID: 10699948
-
Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions.Oncogene. 2009 Sep 10;28(36):3221-34. doi: 10.1038/onc.2009.183. Epub 2009 Jun 29. Oncogene. 2009. PMID: 19561642
-
Lipid rafts and Fas/CD95 signaling in cancer chemotherapy.Recent Pat Anticancer Drug Discov. 2011 Sep;6(3):274-83. doi: 10.2174/157489211796957766. Recent Pat Anticancer Drug Discov. 2011. PMID: 21762074 Review.
Cited by
-
Life in the Fas lane: differential outcomes of Fas signaling.Cell Mol Life Sci. 2013 Nov;70(21):4085-99. doi: 10.1007/s00018-013-1327-z. Epub 2013 Apr 12. Cell Mol Life Sci. 2013. PMID: 23579628 Free PMC article. Review.
-
Multiplicity of Glycosphingolipid-Enriched Microdomain-Driven Immune Signaling.Int J Mol Sci. 2021 Sep 3;22(17):9565. doi: 10.3390/ijms22179565. Int J Mol Sci. 2021. PMID: 34502474 Free PMC article. Review.
-
Caenorhabditis elegans as a platform to study the mechanism of action of synthetic antitumor lipids.Cell Cycle. 2014;13(21):3375-89. doi: 10.4161/15384101.2014.952183. Cell Cycle. 2014. PMID: 25485582 Free PMC article.
-
Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer.Cancers (Basel). 2021 Aug 19;13(16):4173. doi: 10.3390/cancers13164173. Cancers (Basel). 2021. PMID: 34439330 Free PMC article. Review.
-
Inhibition of Granulomatous Inflammation and Prophylactic Treatment of Schistosomiasis with a Combination of Edelfosine and Praziquantel.PLoS Negl Trop Dis. 2015 Jul 20;9(7):e0003893. doi: 10.1371/journal.pntd.0003893. eCollection 2015. PLoS Negl Trop Dis. 2015. PMID: 26191954 Free PMC article.
References
-
- Munder, P.G., and O. Westphal. 1990. Antitumoral and other biomedical activities of synthetic ether lysophospholipids. Chem. Immunol. 49:206–235. - PubMed
-
- Houlihan, W.J., M. Lohmeyer, P. Workman, and S.H. Cheon. 1995. Phospholipid antitumor agents. Med. Res. Rev. 15:157–223. - PubMed
-
- Gajate, C., and F. Mollinedo. 2002. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH(3) (edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab. 3:491–525. - PubMed
-
- Jendrossek, V., and R. Handrick. 2003. Membrane targeted anticancer drugs: potent inducers of apoptosis and putative radiosensitisers. Curr. Med. Chem. Anti-Canc. Agents. 3:343–353. - PubMed
-
- Mollinedo, F., J.L. Fernandez-Luna, C. Gajate, B. Martin-Martin, A. Benito, R. Martinez-Dalmau, and M. Modolell. 1997. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (edelfosine): molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-X(L). Cancer Res. 57:1320–1328. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
