

Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases
- PMID: 15289505
- PMCID: PMC2211976
- DOI: 10.1084/jem.20040435
Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases
Abstract
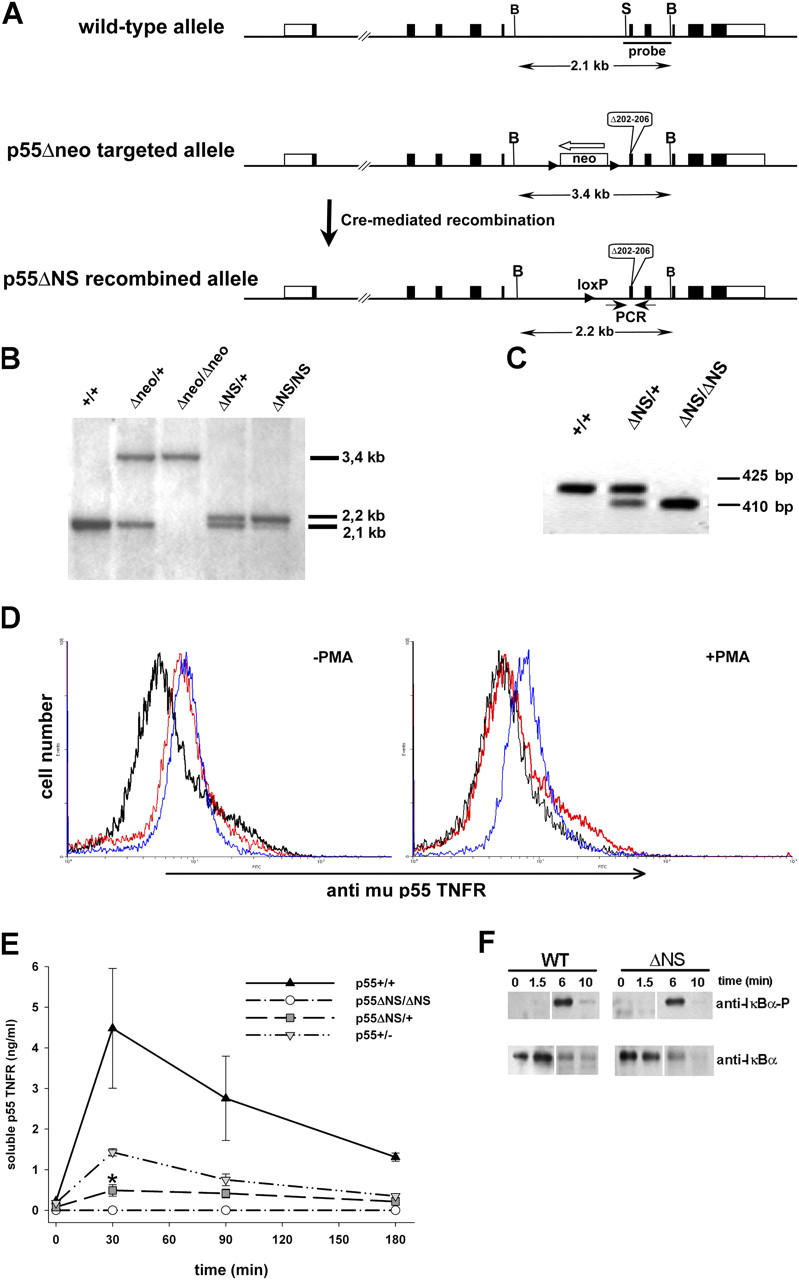
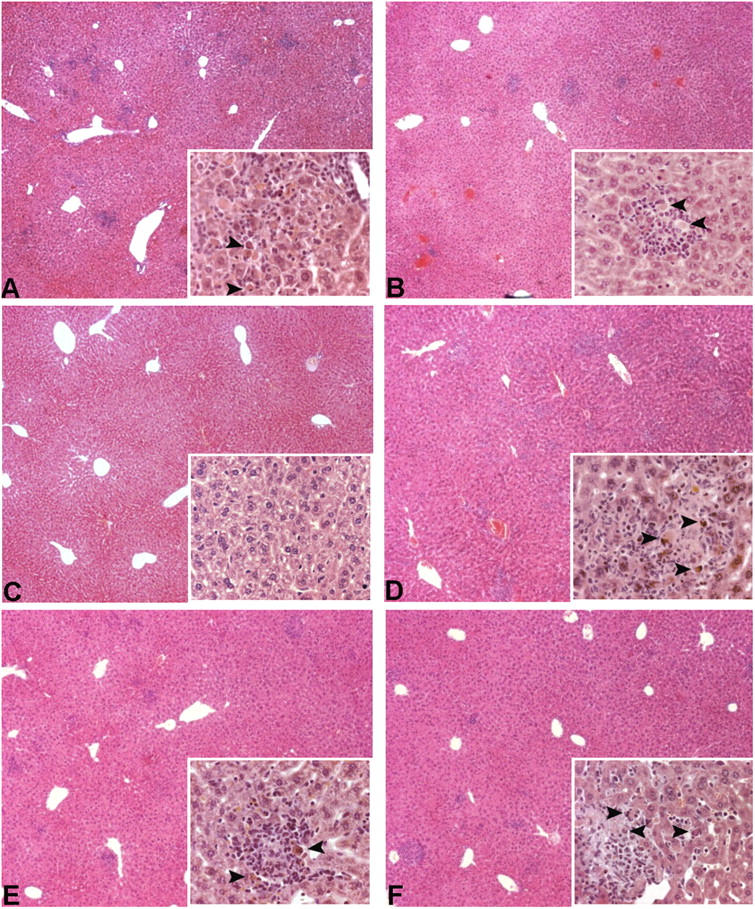
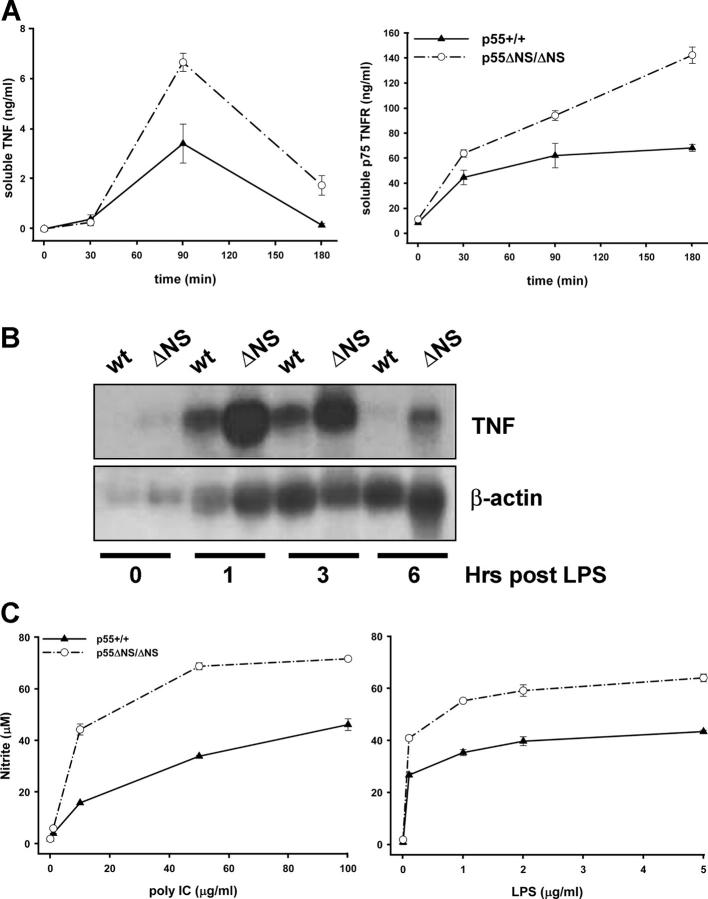
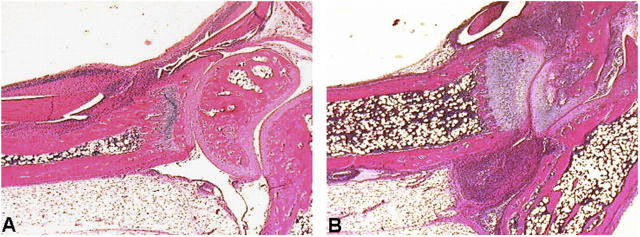
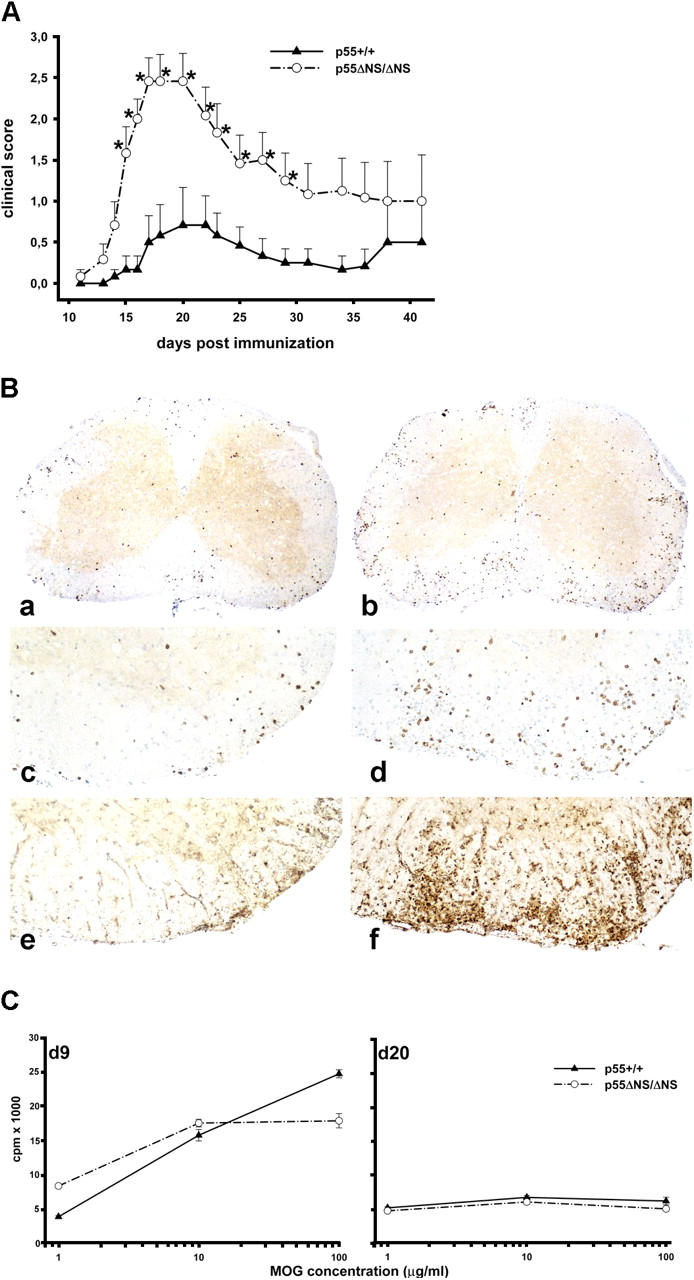
Tumor necrosis factor (TNF) is a potent cytokine exerting critical functions in the activation and regulation of immune and inflammatory responses. Due to its pleiotropic activities, the amplitude and duration of TNF function must be tightly regulated. One of the mechanisms that may have evolved to modulate TNF function is the proteolytic cleavage of its cell surface receptors. In humans, mutations affecting shedding of the p55TNF receptor (R) have been linked with the development of the TNFR-associated periodic syndromes, disorders characterized by recurrent fever attacks and localized inflammation. Here we show that knock-in mice expressing a mutated nonsheddable p55TNFR develop Toll-like receptor-dependent innate immune hyperreactivity, which renders their immune system more efficient at controlling intracellular bacterial infections. Notably, gain of function for antibacterial host defenses ensues at the cost of disbalanced inflammatory reactions that lead to pathology. Mutant mice exhibit spontaneous hepatitis, enhanced susceptibility to endotoxic shock, exacerbated TNF-dependent arthritis, and experimental autoimmune encephalomyelitis. These results introduce a new concept for receptor shedding as a mechanism setting up thresholds of cytokine function to balance resistance and susceptibility to disease. Assessment of p55TNFR shedding may thus be of prognostic value in infectious, inflammatory, and autoimmune diseases.
Figures
References
-
- Smith, C.A., T. Farrah, and R.G. Goodwin. 1994. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 76:959–962. - PubMed
-
- Wallach, D., E.E. Varfolomeev, N.L. Malinin, Y.V. Goltsev, A.V. Kovalenko, and M.P. Boldin. 1999. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 17:331–367. - PubMed
-
- Wajant, H., K. Pfizenmaier, and P. Scheurich. 2003. Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. - PubMed
-
- Pasparakis, M., S. Kousteni, J. Peschon, and G. Kollias. 2000. Tumor necrosis factor and the p55TNF receptor are required for optimal development of the marginal sinus and for migration of follicular dendritic cell precursors into splenic follicles. Cell. Immunol. 201:33–41. - PubMed
-
- Steinshamn, S., M.H. Bemelmans, L.J. van Tits, K. Bergh, W.A. Buurman, and A. Waage. 1996. TNF receptors in murine Candida albicans infection: evidence for an important role of TNF receptor p55 in antifungal defense. J. Immunol. 157:2155–2159. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
