

High genetic diversity of the attachment (G) protein of human metapneumovirus
- PMID: 15297475
- PMCID: PMC497604
- DOI: 10.1128/JCM.42.8.3406-3414.2004
High genetic diversity of the attachment (G) protein of human metapneumovirus
Abstract
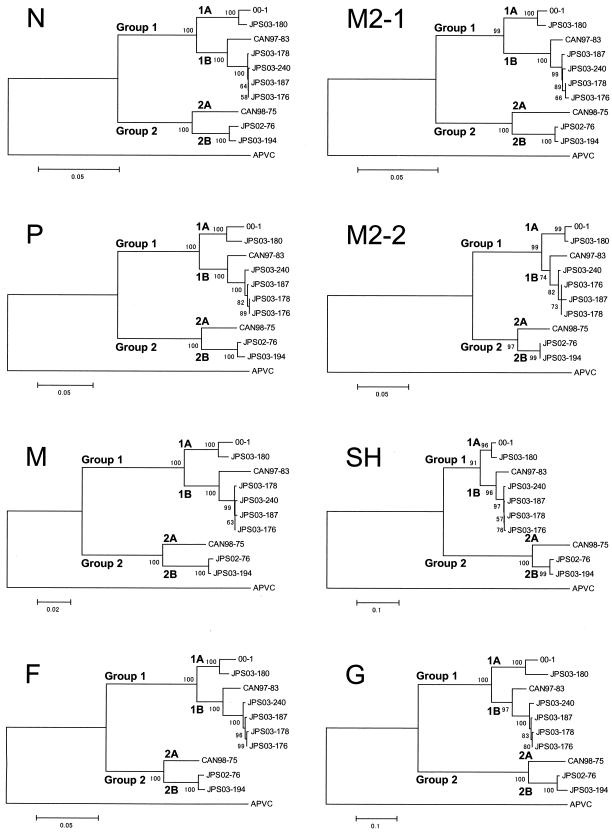
Complete genes encoding the predicted nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), M2-1protein, M2-2protein, small hydrophobic protein (SH), and attachmentprotein (G) of seven newly isolated human metapneumoviruses (hMPVs) were analyzed and compared with previously published data for hMPV genes. Phylogenetic analysis of the nucleotide sequences indicated that there were two genetic groups, tentatively named groups 1 and 2, similar to the grouping of human respiratory syncytial virus. Although the predicted amino acid sequences of N, P, M, F, and M2 were highly conserved between the two groups (amino acid identities, 96% for N, 85% for P, 97% for M, 94% for F, 95% for M2-1, and 90% for M2-2), the amino acid identities of the SH and G proteins were low (SH, 58%; G, 33%). Furthermore, each group could be subdivided into two subgroups by phylogenetic analysis, tentatively named subgroups 1A and 1B and subgroups 2A and 2B. The predicted amino acid sequences of G within members of each subgroup were highly conserved (amino acid identities, 88% for group 1A, 93% for group 1B, and 96% for group 2B). The G of hMPV is thought to be the major antigenic determinant and to play an important role in the production of neutralizing antibodies. Clarification of the antigenic diversity of G is important for epidemiological analysis and for establishment of strategies to prevent hMPV infection.
Figures



References
-
- Biacchesi, S., M. H. Skiadopoulos, G. Boivin, C. T. Hanson, B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2003. Genetic diversity between human metapneumovirus subgroups. Virology 315:1-9. - PubMed
-
- Boivin, G., Y. Abed, G. Pelletier, L. Ruel, D. Moisan, S. Cote, T. C. Peret, D. D. Erdman, and L. J. Anderson. 2002. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 186:1330-1334. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
