

Types and frequencies of sequencing errors in methyl-filtered and high c0t maize genome survey sequences
- PMID: 15299135
- PMCID: PMC520775
- DOI: 10.1104/pp.104.041640
Types and frequencies of sequencing errors in methyl-filtered and high c0t maize genome survey sequences
Abstract
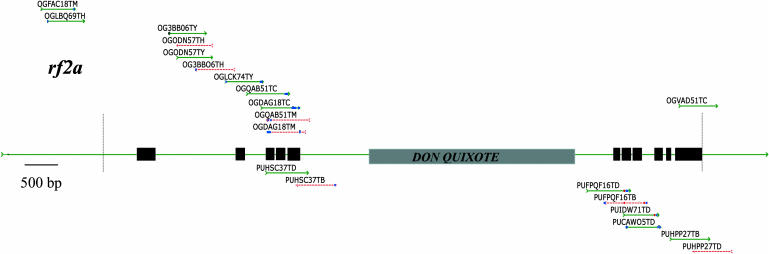
The Maize Genome Sequencing Consortium has deposited into GenBank more than 850,000 maize (Zea mays) genome survey sequences (GSSs) generated via two gene enrichment strategies, methylation filtration and high-C(0)t (HC) fractionation. These GSSs are a valuable resource for generating genome assemblies and the discovery of single nucleotide polymorphisms and nearly identical paralogs. Based on the rate of mismatches between 183 GSSs (105 methylation filtration + 78 HC) and 10 control genes, the rate of sequencing errors in these GSSs is 2.3 x 10(-3). As expected many of these errors were derived from insufficient vector trimming and base-calling errors. Surprisingly, however, some errors were due to cloning artifacts. These G.C to A.T transitions are restricted to HC clones; over 40% of HC clones contain at least one such artifact. Because it is not possible to distinguish the cloning artifacts from biologically relevant polymorphisms, HC sequences should be used with caution for the discovery of single nucleotide polymorphisms or paramorphisms. The average rate of sequencing errors was reduced 6-fold (to 3.6 x 10(-4)) by applying more stringent trimming parameters. This trimming resulted in the loss of only 11% of the bases (15,469/144,968). Due to redundancy among GSSs this more stringent trimming reduced coverage of promoters, exons, and introns by only 0%, 1%, and 4%, respectively. Hence, at the cost of a very modest loss of gene coverage, the quality of these maize GSSs can approach Bermuda standards, even prior to assembly.
Figures
References
-
- Bailey J, Gu Z, Clark R, Reinert K, Samonte R, Schwartz S, Adams M, Myers E, Li P, Eichler E (2002) Recent segmental duplications in the human genome. Science 297: 1003–1007 - PubMed
-
- Brown KR, Weatherdon KL, Galligan CL, Skalski V (2002) A nuclear 3′-5′ exonuclease proofreads for the exonuclease-deficient DNA polymerase alpha. DNA Repair (Amst) 1: 795–810 - PubMed
-
- Chou HH, Holmes MH (2001) DNA sequence quality trimming and vector removal. Bioinformatics 17: 1093–1104 - PubMed
-
- Emrich SJ, Aluru S, Fu Y, Wen TJ, Narayanan M, Guo L, Ashlock D, Schnable PS (2004) A strategy for assembling the maize (Zea mays L.) genome. Bioinformatics 20: 140–147 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
