

Statins inhibit HIV-1 infection by down-regulating Rho activity
- PMID: 15314078
- PMCID: PMC2211926
- DOI: 10.1084/jem.20040061
Statins inhibit HIV-1 infection by down-regulating Rho activity
Abstract
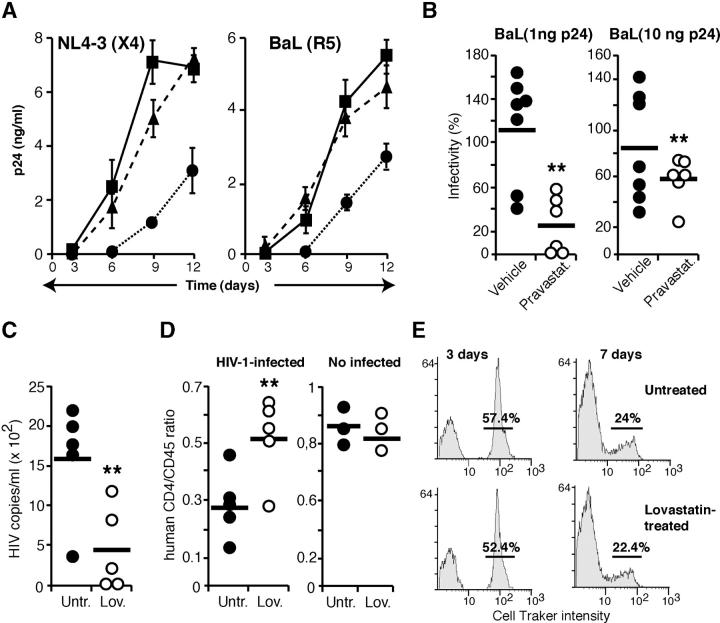
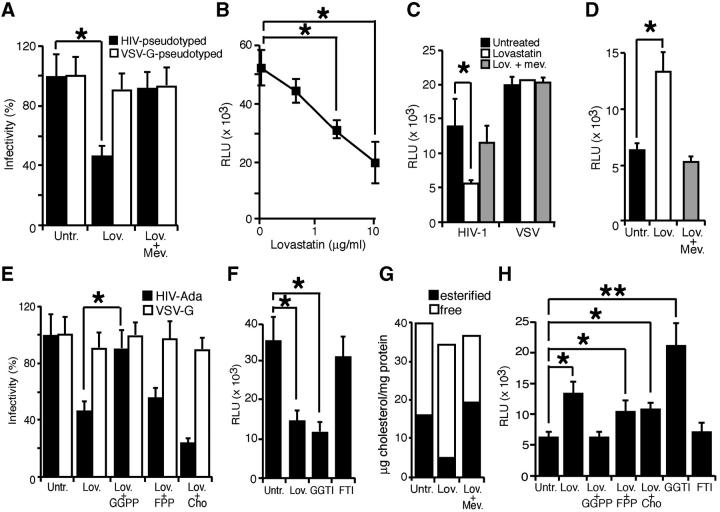
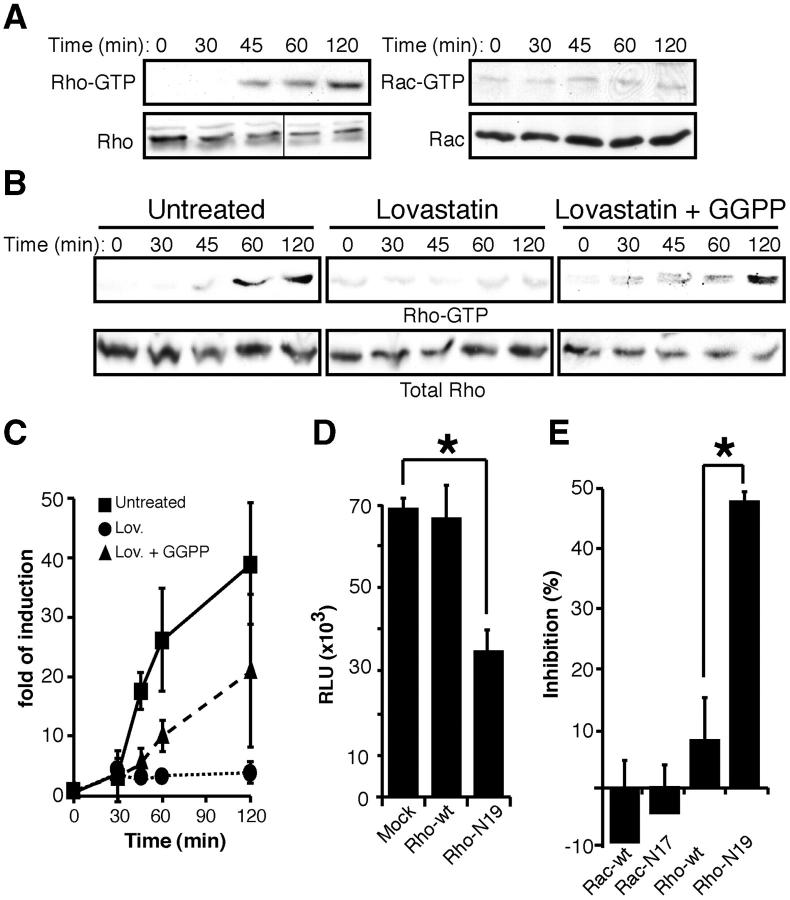
Human immunodeficiency virus (HIV)-1 infectivity requires actin-dependent clustering of host lipid raft-associated receptors, a process that might be linked to Rho guanosine triphosphatase (GTPase) activation. Rho GTPase activity can be negatively regulated by statins, a family of drugs used to treat hypercholesterolemia in man. Statins mediate inhibition of Rho GTPases by impeding prenylation of small G proteins through blockade of 3-hydroxy-3-methylglutaryl coenzyme A reductase. We show that statins decreased viral load and increased CD4+ cell counts in acute infection models and in chronically HIV-1-infected patients. Viral entry and exit was reduced in statin-treated cells, and inhibition was blocked by the addition of l-mevalonate or of geranylgeranylpyrophosphate, but not by cholesterol. Cell treatment with a geranylgeranyl transferase inhibitor, but not a farnesyl transferase inhibitor, specifically inhibited entry of HIV-1-pseudotyped viruses. Statins blocked Rho-A activation induced by HIV-1 binding to target cells, and expression of the dominant negative mutant RhoN19 inhibited HIV-1 envelope fusion with target cell membranes, reducing cell infection rates. We suggest that statins have direct anti-HIV-1 effects by targeting Rho.
Figures
References
-
- Mañes, S., G. del Real, and C. Martínez-A. 2003. Pathogens: raft hijackers. Nat. Rev. Immunol. 3:557–568. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
