

Interactions among human immunodeficiency virus (HIV)-1, interferon-gamma and receptor of activated NF-kappa B ligand (RANKL): implications for HIV pathogenesis
- PMID: 15320903
- PMCID: PMC1809133
- DOI: 10.1111/j.1365-2249.2004.02568.x
Interactions among human immunodeficiency virus (HIV)-1, interferon-gamma and receptor of activated NF-kappa B ligand (RANKL): implications for HIV pathogenesis
Abstract
We reported recently that exposure of human T cells to soluble HIV-1 envelope glycoprotein gp120 induced biologically active tumour necrosis factor (TNF)-alpha-related cytokine receptor of activated NF-kappaB ligand (RANKL), the primary drive to osteoclast differentiation and bone resorption. Furthermore, certain anti-HIV protease inhibitors linked clinically to accelerated bone loss in HIV disease blocked the physiological control of RANKL activity by interferon (IFN)-gamma through inhibition of degradation of the RANKL nuclear adapter signalling protein, TNF receptor associated protein 6 (TRAF6). We now report a series of reciprocal interactions among HIV-1, RANKL and IFN-gamma. RANKL augmented HIV replication in acutely and chronically infected cells of T lymphocyte and monocyte lineage, effects which occurred at a transcriptional level in conjunction with activation of NF-kappaB. TNF-alpha and RANKL were markedly synergistic in induction of HIV. Low pharmacological levels of IFN-gamma (0.75-3 ng/ml) suppressed RANKL-driven enhancement of HIV replication, as did L-T6DP-1, a cell-permeable peptide inhibitor of TRAF6. In contrast, HIV replication induced by TNF-alpha and phorbol ester were not inhibited, and in some cases augmented, by IFN-gamma. We conclude that a positive feedback loop exists between RANKL production and HIV replication, which may be relevant to both the pathophysiology of HIV-linked osteopenia and control of HIV growth. This pathway appears distinct from those of other cytokine activators of HIV, with respect to its utilization of TRAF6 and its suppression by IFN-gamma. These data raise the possibility that TRAF-specific inhibitory peptides, alone or in conjunction with IFN-gamma, could be used to regulate HIV activation in vivo.
Figures
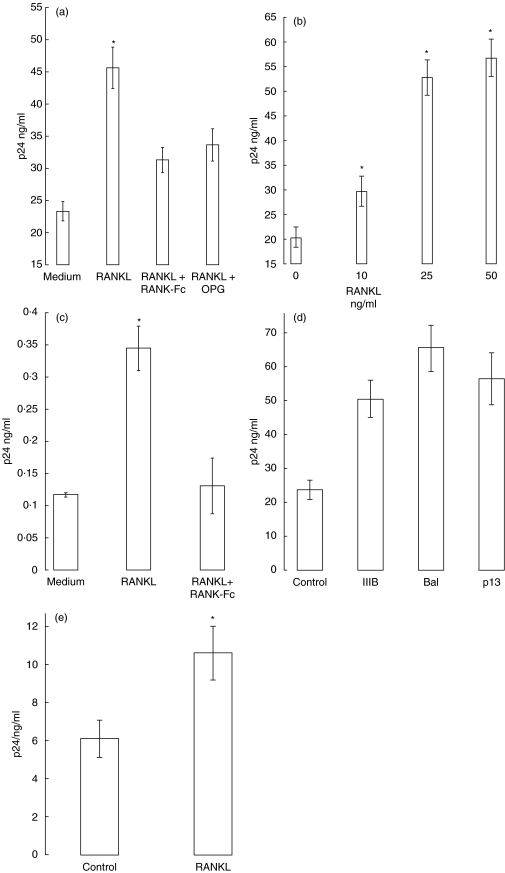
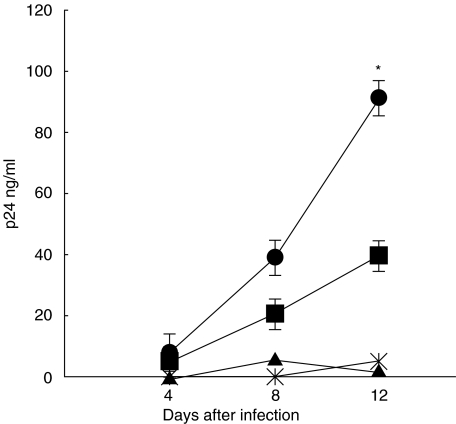
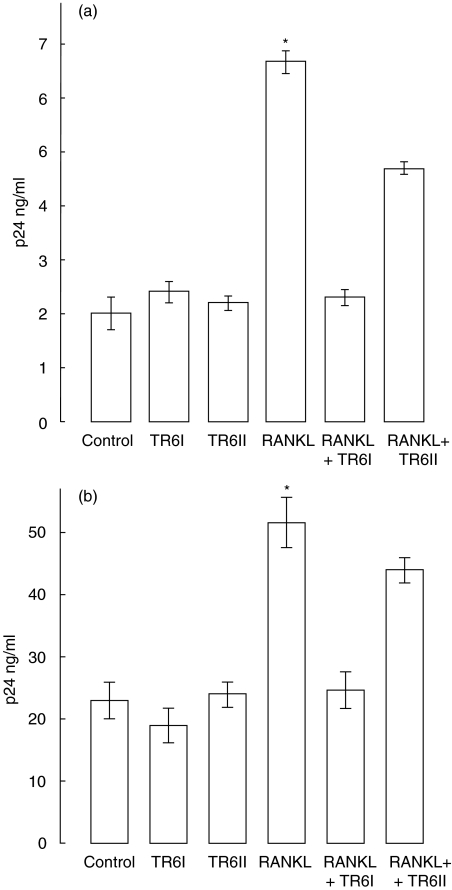
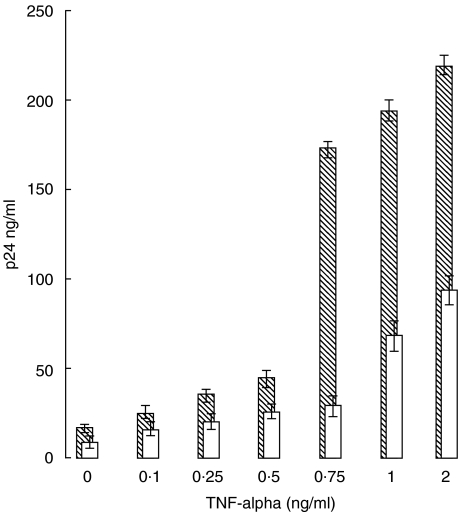
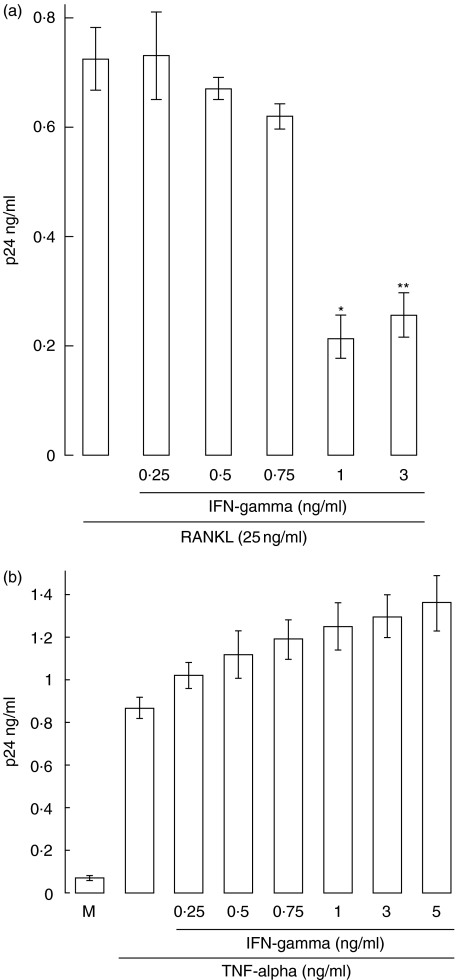
 RANKL alone).
RANKL alone).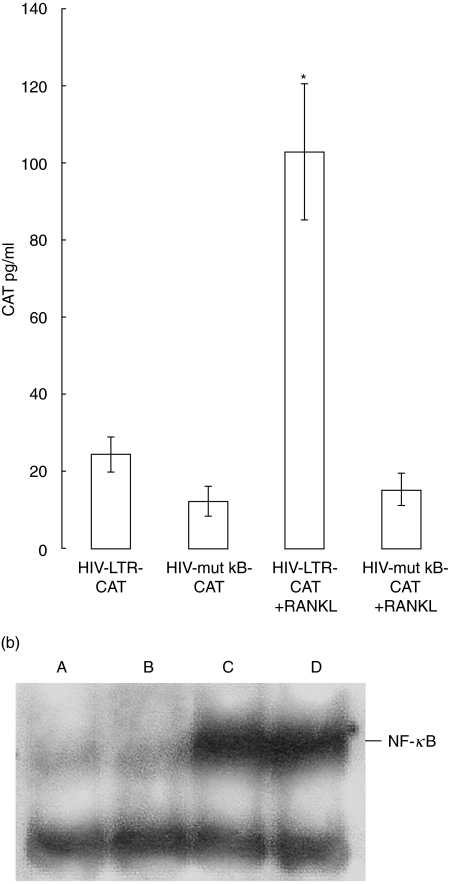
Similar articles
-
T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma.Nature. 2000 Nov 30;408(6812):600-5. doi: 10.1038/35046102. Nature. 2000. PMID: 11117749
-
HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk.J Biol Chem. 2003 Nov 28;278(48):48251-8. doi: 10.1074/jbc.M304676200. Epub 2003 Sep 15. J Biol Chem. 2003. PMID: 12975380
-
Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumor necrosis factor-alpha.Free Radic Biol Med. 2006 May 1;40(9):1483-93. doi: 10.1016/j.freeradbiomed.2005.10.066. Epub 2005 Dec 9. Free Radic Biol Med. 2006. PMID: 16632109
-
Signaling crosstalk between RANKL and interferons in osteoclast differentiation.Arthritis Res. 2002;4 Suppl 3(Suppl 3):S227-32. doi: 10.1186/ar581. Epub 2002 May 9. Arthritis Res. 2002. PMID: 12110142 Free PMC article. Review.
-
A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function.Biochem Biophys Res Commun. 1999 Mar 24;256(3):449-55. doi: 10.1006/bbrc.1999.0252. Biochem Biophys Res Commun. 1999. PMID: 10080918 Review.
Cited by
-
Prevalence and predictors of bone health among perinatally HIV-infected adolescents.AIDS. 2020 Nov 15;34(14):2061-2070. doi: 10.1097/QAD.0000000000002686. AIDS. 2020. PMID: 32910060 Free PMC article.
-
Perturbations of circulating levels of RANKL-osteoprotegerin axis in relation to lipids and progression of atherosclerosis in HIV-infected and -uninfected adults: ACTG NWCS 332/A5078 Study.AIDS Res Hum Retroviruses. 2013 Jun;29(6):938-48. doi: 10.1089/AID.2012.0305. Epub 2013 Feb 25. AIDS Res Hum Retroviruses. 2013. PMID: 23351153 Free PMC article.
-
Endocrinological aspects of HIV infection.J Endocrinol Invest. 2018 Aug;41(8):881-899. doi: 10.1007/s40618-017-0812-x. Epub 2018 Jan 8. J Endocrinol Invest. 2018. PMID: 29313284 Review.
-
Modulation of osteoclastogenesis induced by nucleoside reverse transcriptase inhibitors.AIDS Res Hum Retroviruses. 2006 Nov;22(11):1131-41. doi: 10.1089/aid.2006.22.1131. AIDS Res Hum Retroviruses. 2006. PMID: 17147500 Free PMC article.
-
Bone mineral density in children and adolescents with perinatal HIV infection.AIDS. 2013 Jan 14;27(2):211-20. doi: 10.1097/QAD.0b013e32835a9b80. AIDS. 2013. PMID: 23032412 Free PMC article.
References
-
- Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9. - PubMed
-
- Fakruddin JM, Laurence J. HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor κB ligand (RANKL) secretion and its modulation by HIV protease inhibitors through interferon-γ/RANKL cross-talk. J Biol Chem. 2003;278:48251–8. - PubMed
-
- Fakruddin JM, Laurence J. Regulation of RANKL expression in HIV infection and its therapy: relationship to HIV-linked osteopenia; XV International AIDS Conference, Bankok, Thailand. 2004. [Abstract WePpB2063].
-
- Knobel H, Guelar A, Vallecillo G, Nogues A. Osteopenia in HIV-infected patients: is it the disease or its treatment? AIDS. 2001;15:807–8. - PubMed
-
- Thomas J, Doherty SM. HIV infection − a risk factor for osteoporosis. J Acquir Immune Defic Syndr. 2003;33:281–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
