

Granulocyte-macrophage colony-stimulating factor enhancer activation requires cooperation between NFAT and AP-1 elements and is associated with extensive nucleosome reorganization
- PMID: 15340054
- PMCID: PMC515070
- DOI: 10.1128/MCB.24.18.7914-7930.2004
Granulocyte-macrophage colony-stimulating factor enhancer activation requires cooperation between NFAT and AP-1 elements and is associated with extensive nucleosome reorganization
Abstract
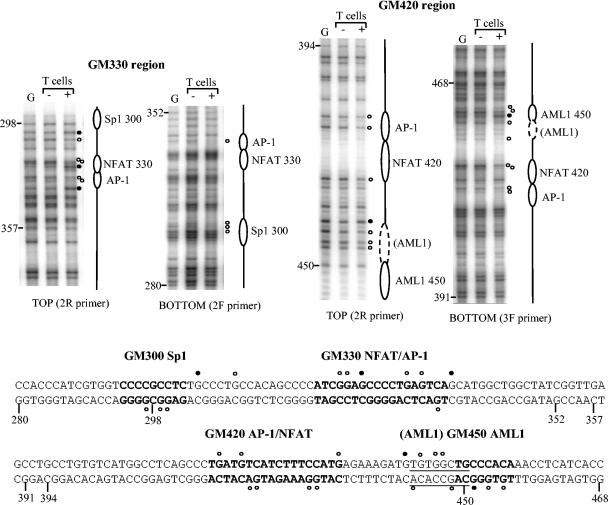
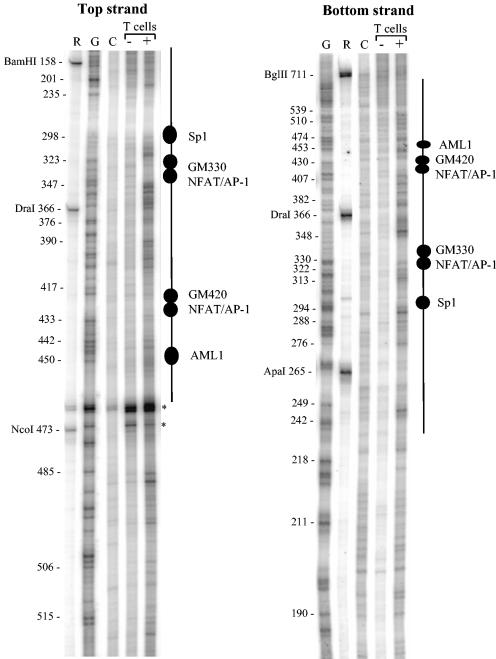
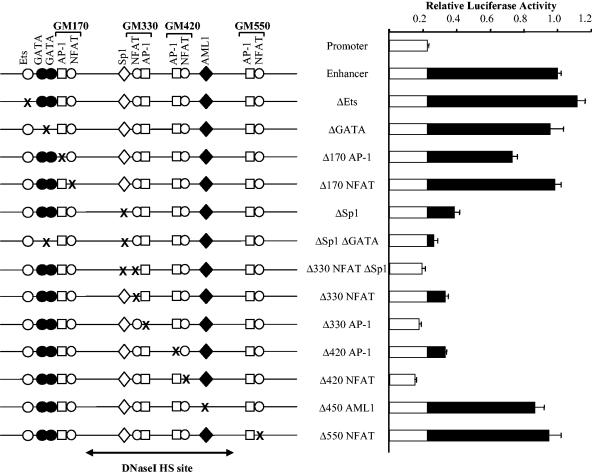
The human granulocyte-macrophage colony-stimulating factor (GM-CSF) gene is activated by an NFAT-dependent enhancer forming an inducible DNase I hypersensitive (DH) site. The enhancer core comprising the DH site contains the GM330 and GM420 elements that bind NFAT and AP-1 cooperatively. Here we demonstrate that both elements are essential for enhancer activity and that Sp1 and AML1 sites in the enhancer become occupied in vivo only after activation. Chromatin structure analysis revealed that the GM-CSF enhancer core elements are divided between two adjacent nucleosomes that become destabilized and highly accessible after activation. Inducible chromatin reorganization was not restricted to the enhancer core but extended across a 3-kb domain of mobilized nucleosomes, within which the nucleosome repeat length was compressed from approximately 185 to 150 bp. The GM420 element is a high-affinity site that binds NFAT independently of AP-1 but depends on the linked AP-1 site for enhancer function. Nevertheless, just the NFAT motif from the GM420 element was sufficient to form a DH site within chromatin even in the absence of the AP-1 site. Hence, NFAT has the potential to cooperate with other transcription factors by promoting chromatin remodelling and increasing accessibility at inducible regulatory elements.
Figures







Similar articles
-
Human granulocyte-macrophage colony-stimulating factor enhancer function is associated with cooperative interactions between AP-1 and NFATp/c.Mol Cell Biol. 1995 Apr;15(4):2071-9. doi: 10.1128/MCB.15.4.2071. Mol Cell Biol. 1995. PMID: 7891702 Free PMC article.
-
Multiple signals are required for function of the human granulocyte-macrophage colony-stimulating factor gene promoter in T cells.J Immunol. 1995 Aug 1;155(3):1240-51. J Immunol. 1995. PMID: 7636192
-
Regulation of GM-CSF gene transcription by core-binding factor.Cell Growth Differ. 1996 Jul;7(7):917-22. Cell Growth Differ. 1996. PMID: 8809409
-
Mechanisms of transcriptional regulation of the human IL-3/GM-CSF locus by inducible tissue-specific promoters and enhancers.Crit Rev Immunol. 2004;24(6):385-408. doi: 10.1615/critrevimmunol.v24.i6.10. Crit Rev Immunol. 2004. PMID: 15777160 Review.
-
Signals for activation of the GM-CSF promoter and enhancer in T cells.Crit Rev Immunol. 1997;17(3-4):301-23. doi: 10.1615/critrevimmunol.v17.i3-4.30. Crit Rev Immunol. 1997. PMID: 9202885 Review.
Cited by
-
Inhibitory effects of 405 nm irradiation on Chlamydia trachomatis growth and characterization of the ensuing inflammatory response in HeLa cells.BMC Microbiol. 2012 Aug 15;12:176. doi: 10.1186/1471-2180-12-176. BMC Microbiol. 2012. PMID: 22894815 Free PMC article.
-
Nuclear factor of activated T cells 1 (NFAT1)-induced permissive chromatin modification facilitates nuclear factor-κB (NF-κB)-mediated interleukin-9 (IL-9) transactivation.J Biol Chem. 2012 May 4;287(19):15445-57. doi: 10.1074/jbc.M112.340356. Epub 2012 Mar 15. J Biol Chem. 2012. PMID: 22427656 Free PMC article.
-
Modulation of NFAT-dependent gene expression by the RhoA signaling pathway in T cells.J Leukoc Biol. 2007 Aug;82(2):361-9. doi: 10.1189/jlb.0206120. Epub 2007 May 14. J Leukoc Biol. 2007. PMID: 17502338 Free PMC article.
-
RUNX inhibitor suppresses graft-versus-host disease through targeting RUNX-NFATC2 axis.EJHaem. 2021 May 19;2(3):449-458. doi: 10.1002/jha2.230. eCollection 2021 Aug. EJHaem. 2021. PMID: 35844683 Free PMC article.
-
Receptor Signaling Directs Global Recruitment of Pre-existing Transcription Factors to Inducible Elements.Yale J Biol Med. 2016 Dec 23;89(4):591-596. eCollection 2016 Dec. Yale J Biol Med. 2016. PMID: 28018147 Free PMC article. Review.
References
-
- Avots, A., M. Buttmann, S. Chuvpilo, C. Escher, U. Smola, A. J. Bannister, U. R. Rapp, T. Kouzarides, and E. Serfling. 1999. CBP/p300 integrates Raf/Rac-signaling pathways in the transcriptional induction of NF-ATc during T cell activation. Immunity 10:515-524. - PubMed
-
- Bert, A. G., J. Burrows, A. Hawwari, M. A. Vadas, and P. N. Cockerill. 2000. Reconstitution of T cell-specific transcription directed by composite NFAT/Oct elements. J. Immunol. 165:5646-5655. - PubMed
-
- Bert, A. G., J. Burrows, C. S. Osborne, and P. N. Cockerill. 2000. Generation of an improved luciferase reporter gene plasmid that employs a novel mechanism for high copy replication. Plasmid 44:173-182. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Research Materials