

Transcriptional control in the segmentation gene network of Drosophila
- PMID: 15340490
- PMCID: PMC514885
- DOI: 10.1371/journal.pbio.0020271
Transcriptional control in the segmentation gene network of Drosophila
Abstract
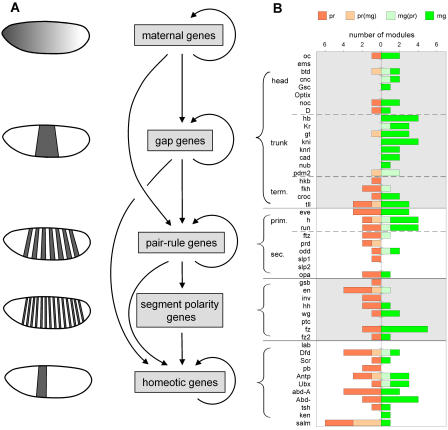
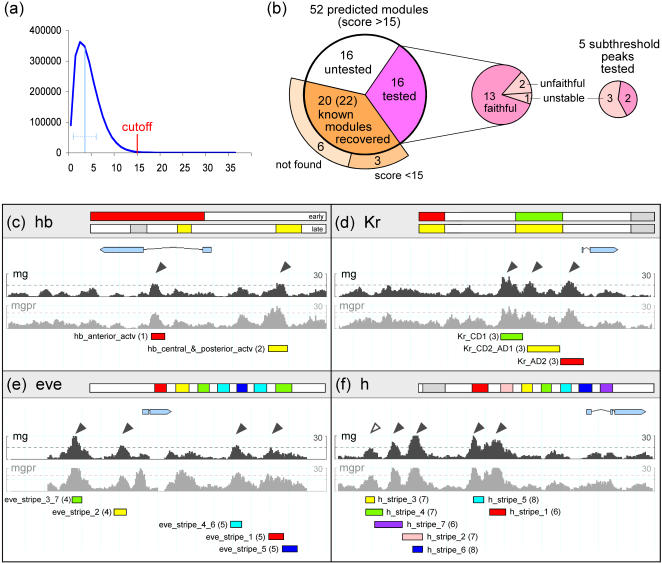
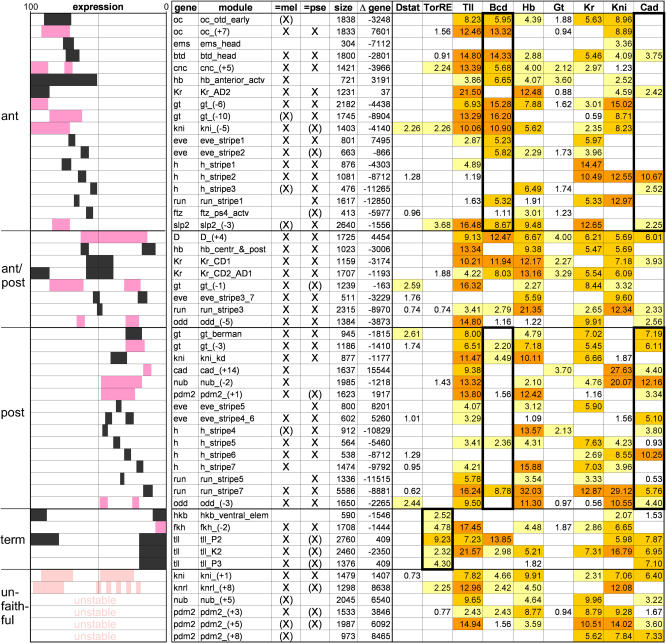
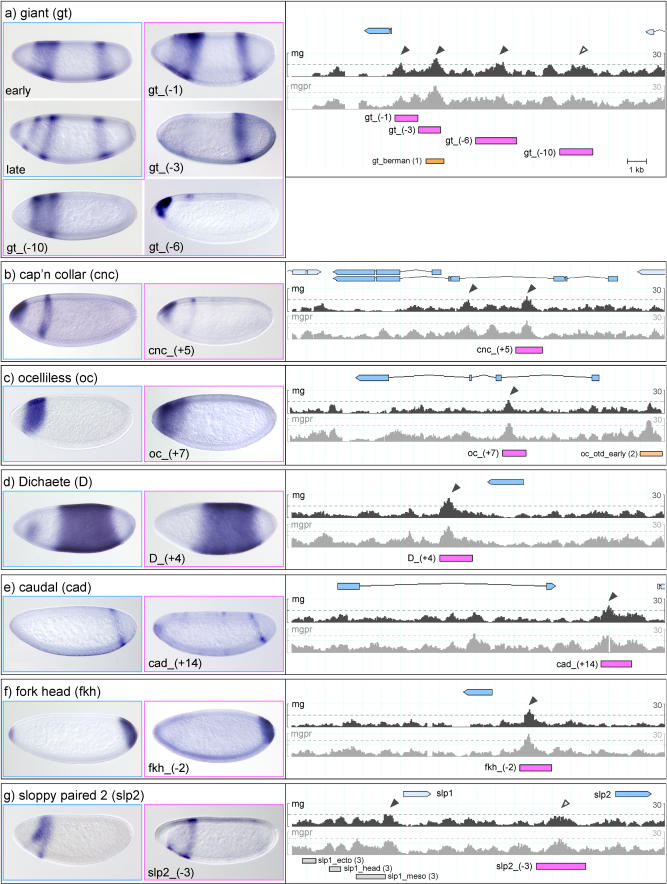
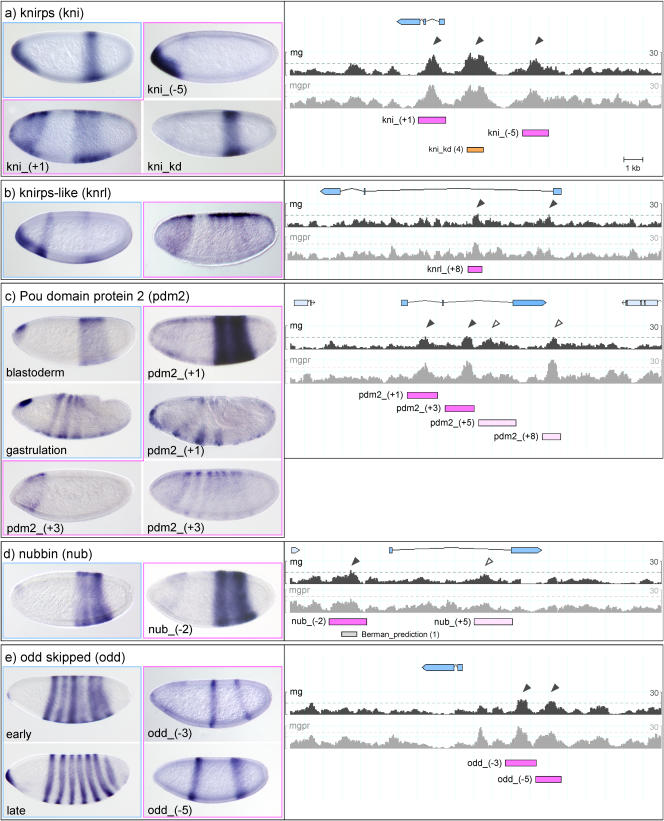
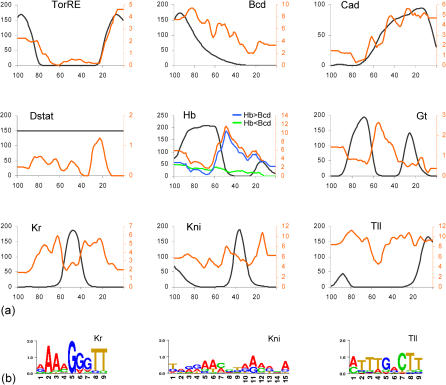
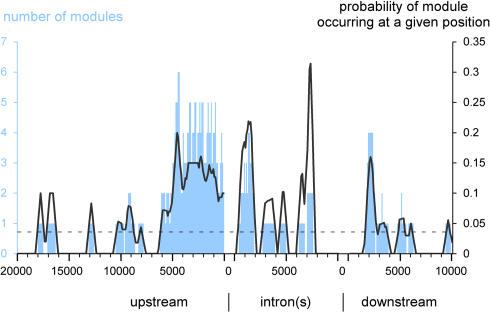
The segmentation gene network of Drosophila consists of maternal and zygotic factors that generate, by transcriptional (cross-) regulation, expression patterns of increasing complexity along the anterior-posterior axis of the embryo. Using known binding site information for maternal and zygotic gap transcription factors, the computer algorithm Ahab recovers known segmentation control elements (modules) with excellent success and predicts many novel modules within the network and genome-wide. We show that novel module predictions are highly enriched in the network and typically clustered proximal to the promoter, not only upstream, but also in intronic space and downstream. When placed upstream of a reporter gene, they consistently drive patterned blastoderm expression, in most cases faithfully producing one or more pattern elements of the endogenous gene. Moreover, we demonstrate for the entire set of known and newly validated modules that Ahab's prediction of binding sites correlates well with the expression patterns produced by the modules, revealing basic rules governing their composition. Specifically, we show that maternal factors consistently act as activators and that gap factors act as repressors, except for the bimodal factor Hunchback. Our data suggest a simple context-dependent rule for its switch from repressive to activating function. Overall, the composition of modules appears well fitted to the spatiotemporal distribution of their positive and negative input factors. Finally, by comparing Ahab predictions with different categories of transcription factor input, we confirm the global regulatory structure of the segmentation gene network, but find odd skipped behaving like a primary pair-rule gene. The study expands our knowledge of the segmentation gene network by increasing the number of experimentally tested modules by 50%. For the first time, the entire set of validated modules is analyzed for binding site composition under a uniform set of criteria, permitting the definition of basic composition rules. The study demonstrates that computational methods are a powerful complement to experimental approaches in the analysis of transcription networks.
Figures
References
-
- Akam M. The molecular basis for metameric pattern in the Drosophila embryo. Development. 1987;101:1–22. - PubMed
-
- Arnone MI, Davidson EH. The hardwiring of development: Organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. - PubMed
-
- Arnosti DN. Analysis and function of transcriptional regulatory elements: Insights from Drosophila . Annu Rev Entomol. 2003;48:579–602. - PubMed
-
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193:723–750. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
