

Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: assessment of correlated reaction subsets that comprise network states
- PMID: 15342562
- PMCID: PMC515326
- DOI: 10.1101/gr.2546004
Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: assessment of correlated reaction subsets that comprise network states
Abstract
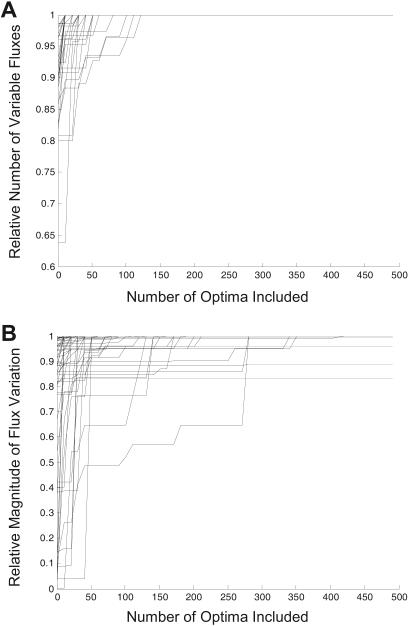
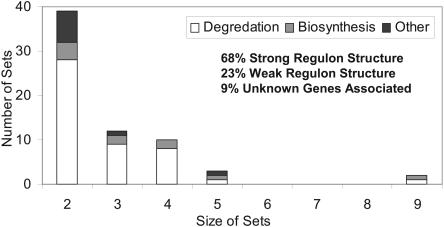
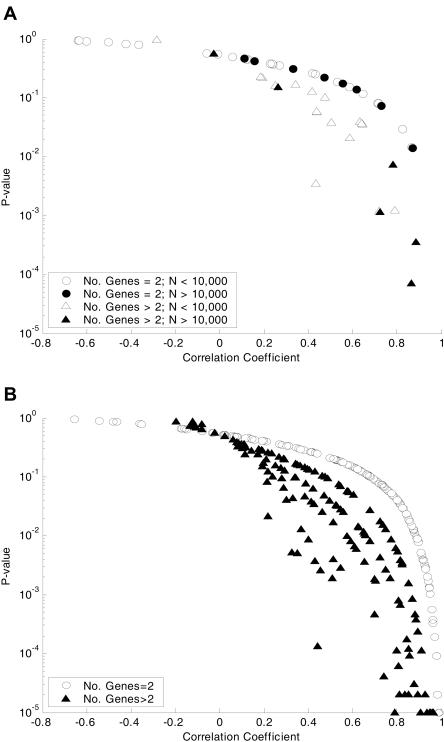
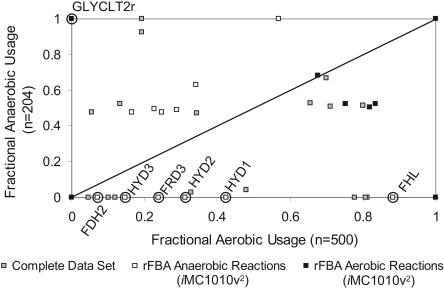
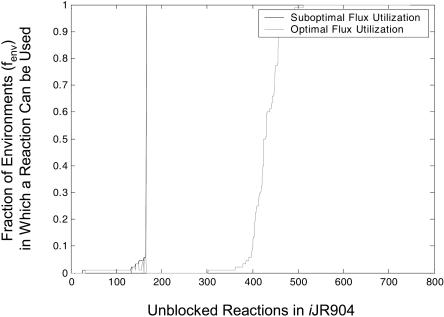
The constraint-based analysis of genome-scale metabolic and regulatory networks has been successful in predicting phenotypes and useful for analyzing high-throughput data sets. Within this modeling framework, linear optimization has been used to study genome-scale metabolic models, resulting in the enumeration of single optimal solutions describing the best use of the network to support growth. Here mixed-integer linear programming was used to calculate and study a subset of the alternate optimal solutions for a genome-scale metabolic model of Escherichia coli (iJR904) under a wide variety of environmental conditions. Analysis of the calculated sets of optimal solutions found that: (1) only a small subset of reactions in the network have variable fluxes across optima; (2) sets of reactions that are always used together in optimal solutions, correlated reaction sets, showed moderate agreement with the currently known transcriptional regulatory structure in E. coli and available expression data, and (3) reactions that are used under certain environmental conditions can provide clues about network regulatory needs. In addition, calculation of suboptimal flux distributions, using flux variability analysis, identified reactions which are used under significantly more environmental conditions suboptimally than optimally. Together these results demonstrate the utilization of reactions in genome-scale models under a variety of different growth conditions.
Figures

References
-
- Almaas, E., Kovacs, B., Vicsek, T., Oltvai, Z.N., and Barabasi, A.L. 2004. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427: 839-843. - PubMed
-
- Burgard, A.P. and Maranas, C.D. 2001. Probing the performance limits of the Escherichia coli metabolic network subject to gene additions or deletions. Biotechnol. Bioeng. 74: 364-375. - PubMed
-
- Burgard, A.P., Vaidyaraman, S., and Maranas, C.D. 2001. Minimal reaction sets for Escherichia coli metabolism under different growth requirements and uptake environments. Biotechnol. Prog. 17: 791-797. - PubMed
WEB SITE REFERENCES
-
- https://asap.ahabs.wisc.edu/~glasner/Protocols/DataDefinitionDefinitions...; Web site describes how the estimated transcript copy number is calculated from the gene expression data.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources