

Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5
- PMID: 15367659
- PMCID: PMC516754
- DOI: 10.1128/MCB.24.19.8374-8385.2004
Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5
Abstract
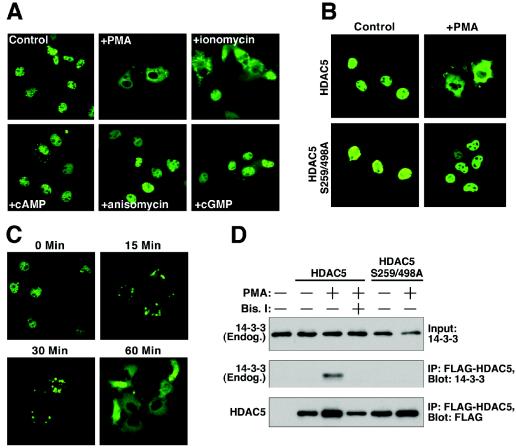
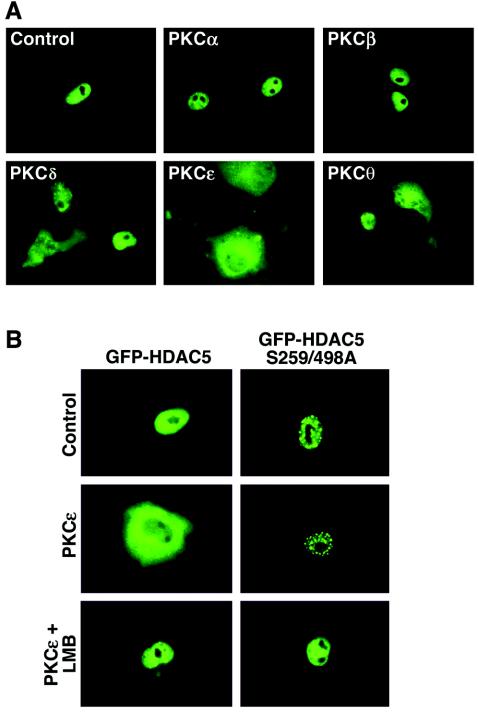
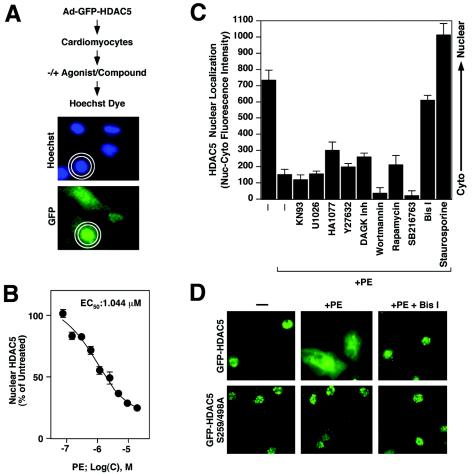
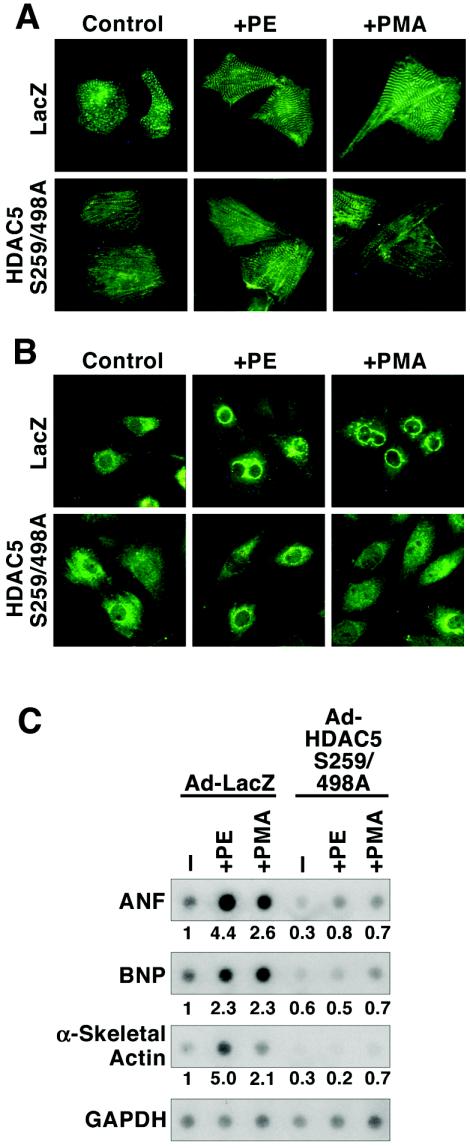
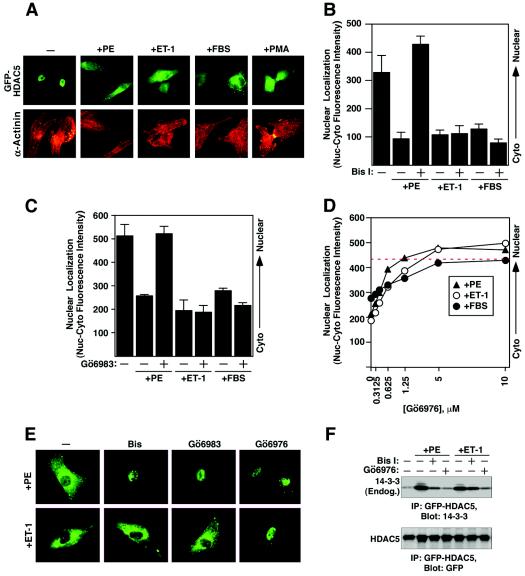
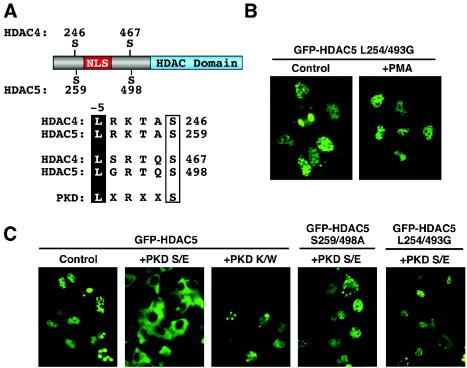
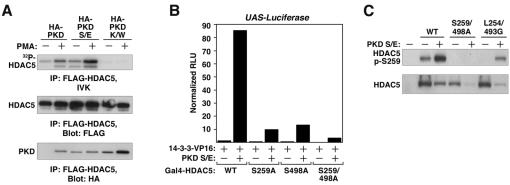
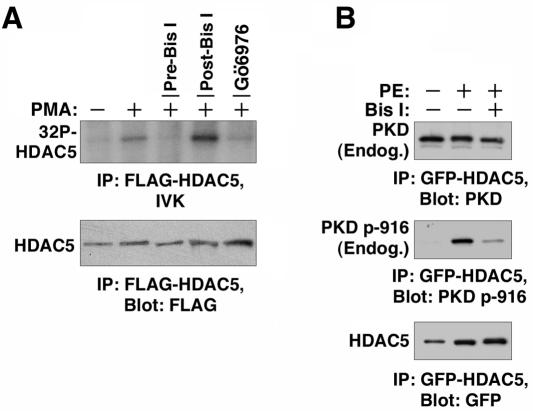
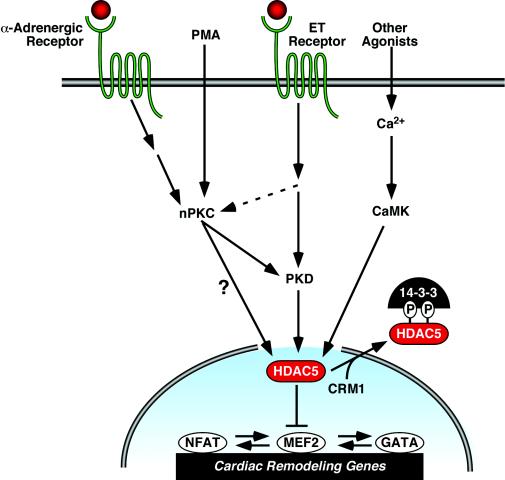
A variety of stress signals stimulate cardiac myocytes to undergo hypertrophy. Persistent cardiac hypertrophy is associated with elevated risk for the development of heart failure. Recently, we showed that class II histone deacetylases (HDACs) suppress cardiac hypertrophy and that stress signals neutralize this repressive function by triggering phosphorylation- and CRM1-dependent nuclear export of these chromatin-modifying enzymes. However, the identities of cardiac HDAC kinases have remained unclear. Here, we demonstrate that signaling by protein kinase C (PKC) is sufficient and, in some cases, necessary to drive nuclear export of class II HDAC5 in cardiomyocytes. Inhibition of PKC prevents nucleocytoplasmic shuttling of HDAC5 in response to a subset of hypertrophic agonists. Moreover, a nonphosphorylatable HDAC5 mutant is refractory to PKC signaling and blocks cardiomyocyte hypertrophy mediated by pharmacological activators of PKC. We also demonstrate that protein kinase D (PKD), a downstream effector of PKC, directly phosphorylates HDAC5 and stimulates its nuclear export. These findings reveal a novel function for the PKC/PKD axis in coupling extracellular cues to chromatin modifications that control cellular growth, and they suggest potential utility for small-molecule inhibitors of this pathway in the treatment of pathological cardiac gene expression.
Figures
References
-
- Allo, S. N., L. L. Carl, and H. E. Morgan. 1992. Acceleration of growth of cultured cardiomyocytes and translocation of protein kinase C. Am. J. Physiol. 263:C319-C325. - PubMed
-
- Antos, C. L., T. A. McKinsey, M. Dreitz, L. M. Hollingsworth, C. L. Zhang, K. Schreiber, H. Rindt, R. J. Gorczynski, and E. N. Olson. 2003. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 278:28930-28937. - PubMed
-
- Braz, J. C., K. Gregory, A. Pathak, W. Zhao, B. Sahin, R. Klevitsky, T. F. Kimball, J. N. Lorenz, A. C. Nairn, S. B. Liggett, I. Bodi, S. Wang, A. Schwartz, E. G. Lakatta, A. A. DePaoli-Roach, J. Robbins, T. E. Hewett, J. A. Bibb, M. V. Westfall, E. G. Kranias, and J. D. Molkentin. 2004. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10:248-254. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases