

The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells
- PMID: 15367687
- PMCID: PMC516745
- DOI: 10.1128/MCB.24.19.8691-8704.2004
The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells
Abstract
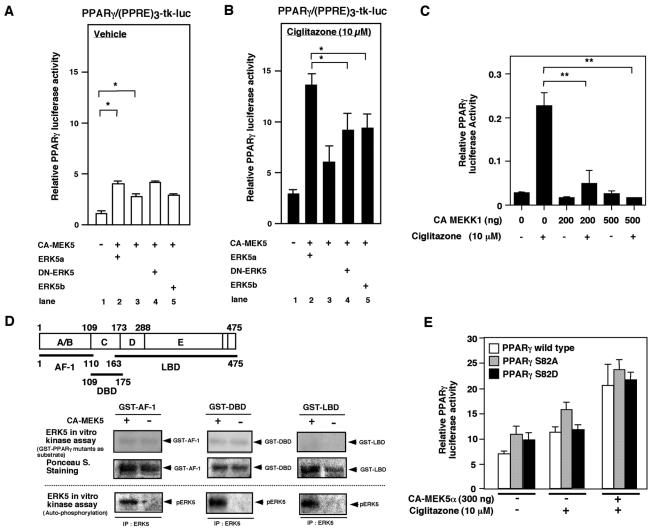
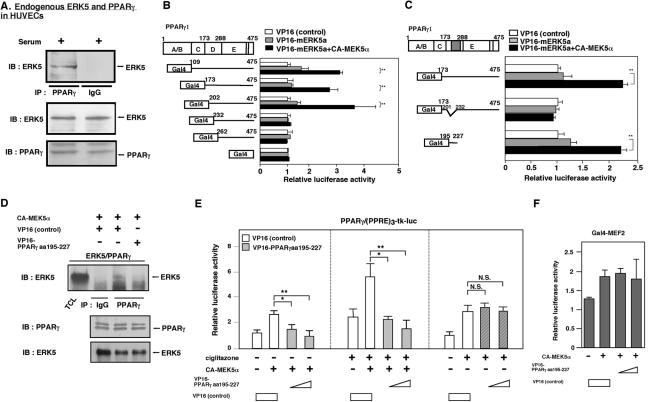
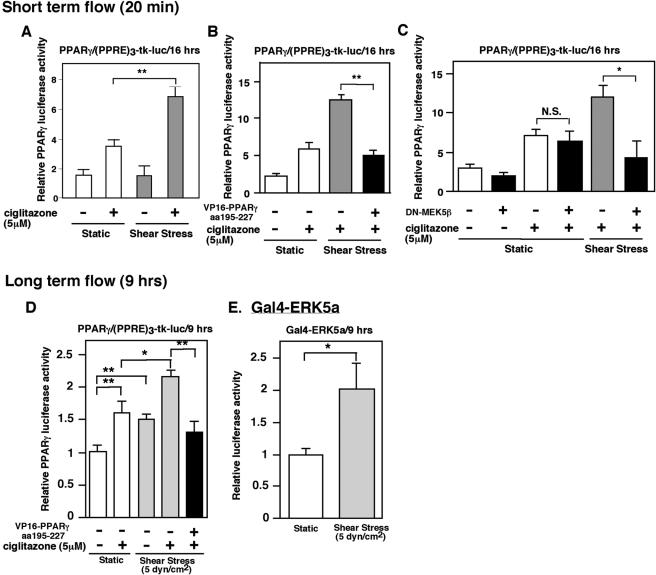
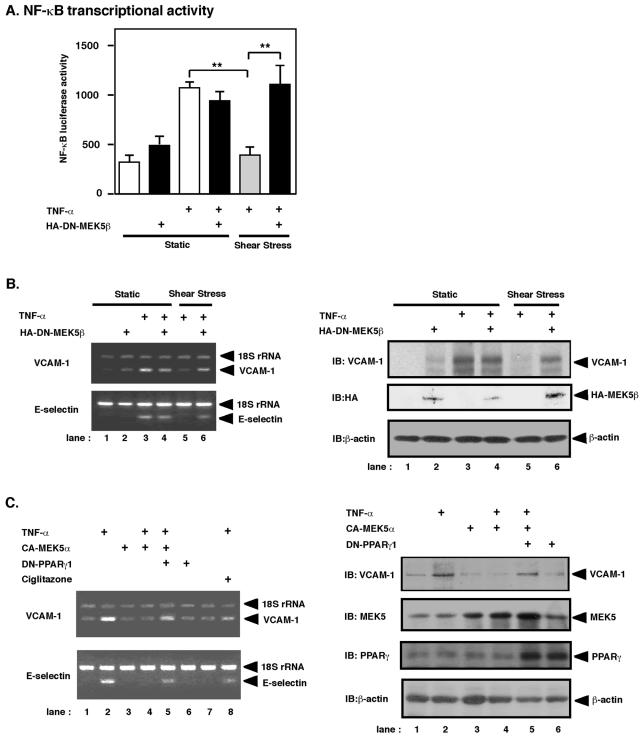
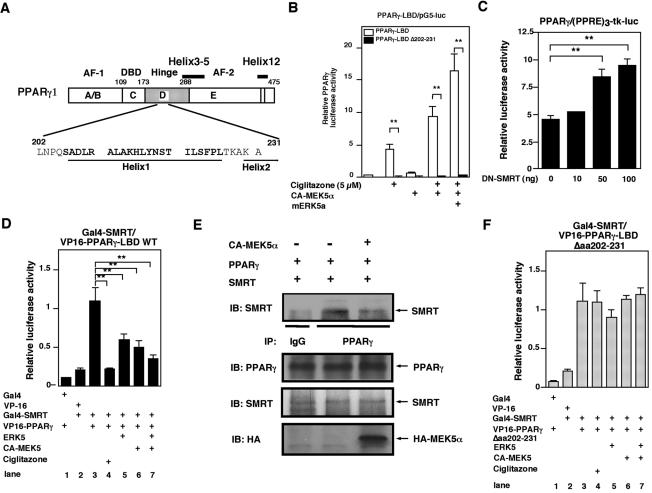
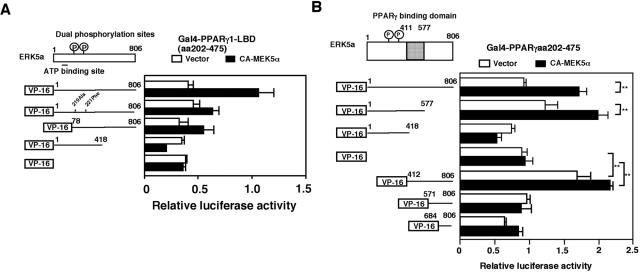
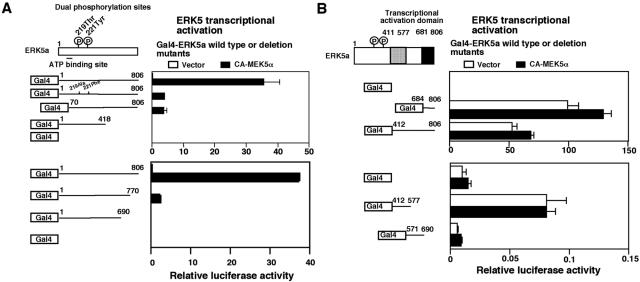
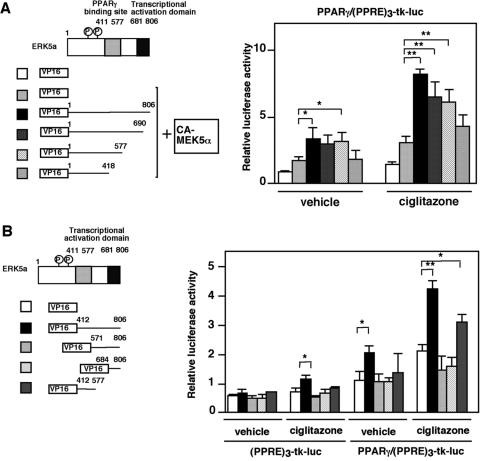
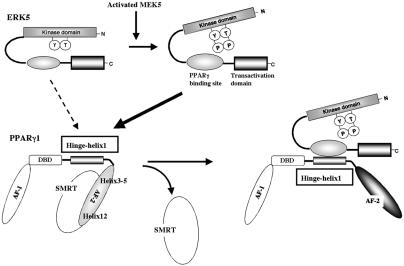
Peroxisome proliferator-activated receptors (PPAR) are ligand-activated transcription factors that form a subfamily of the nuclear receptor gene family. Since both flow and PPARgamma have atheroprotective effects and extracellular signal-regulated kinase 5 (ERK5) kinase activity is significantly increased by flow, we investigated whether ERK5 kinase regulates PPARgamma activity. We found that activation of ERK5 induced PPARgamma1 activation in endothelial cells (ECs). However, we could not detect PPARgamma phosphorylation by incubation with activated ERK5 in vitro, in contrast to ERK1/2 and JNK, suggesting a role for ERK5 as a scaffold. Endogenous PPARgamma1 was coimmunoprecipitated with endogenous ERK5 in ECs. By mammalian two-hybrid analysis, we found that PPARgamma1 associated with ERK5a at the hinge-helix 1 region of PPARgamma1. Expressing a hinge-helix 1 region PPARgamma1 fragment disrupted the ERK5a-PPARgamma1 interaction, suggesting a critical role for hinge-helix 1 region of PPARgamma in the ERK5-PPARgamma interaction. Flow increased ERK5 and PPARgamma1 activation, and the hinge-helix 1 region of the PPARgamma1 fragment and dominant negative MEK5beta significantly reduced flow-induced PPARgamma activation. The dominant negative MEK5beta also prevented flow-mediated inhibition of tumor necrosis factor alpha-mediated NF-kappaB activation and adhesion molecule expression, including vascular cellular adhesion molecule 1 and E-selectin, indicating a physiological role for ERK5 and PPARgamma activation in flow-mediated antiinflammatory effects. We also found that ERK5 kinase activation was required, likely by inducing a conformational change in the NH(2)-terminal region of ERK5 that prevented association of ERK5 and PPARgamma1. Furthermore, association of ERK5a and PPARgamma1 disrupted the interaction of SMRT and PPARgamma1, thereby inducing PPARgamma activation. These data suggest that ERK5 mediates flow- and ligand-induced PPARgamma activation via the interaction of ERK5 with the hinge-helix 1 region of PPARgamma.
Figures
References
-
- Abe, J., M. Kusuhara, R. J. Ulevitch, B. C. Berk, and J. D. Lee. 1996. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 271:16586-16590. - PubMed
-
- Abe, J., M. Takahashi, M. Ishida, J. D. Lee, and B. C. Berk. 1997. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J. Biol. Chem. 272:20389-20394. - PubMed
-
- Aizawa, T., H. Wei, J. M. Miano, J. Abe, B. C. Berk, and C. Yan. 2003. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ. Res. 93:406-413. - PubMed
-
- Ameshima, S., H. Golpon, C. D. Cool, D. Chan, R. W. Vandivier, S. J. Gardai, M. Wick, R. A. Nemenoff, M. W. Geraci, and N. F. Voelkel. 2003. Peroxisome proliferator-activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 92:1162-1169. - PubMed
-
- Cameron, S. J., J. Abe, S. Malik, W. Che, and J. Yang. 2004. Differential role of MEK5α and MEK5β in BMK1/ERK5 activation. J. Biol. Chem. 279:1506-1512. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous