

Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2
- PMID: 15378550
- PMCID: PMC4349519
- DOI: 10.1002/ajmg.a.30251
Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2
Abstract
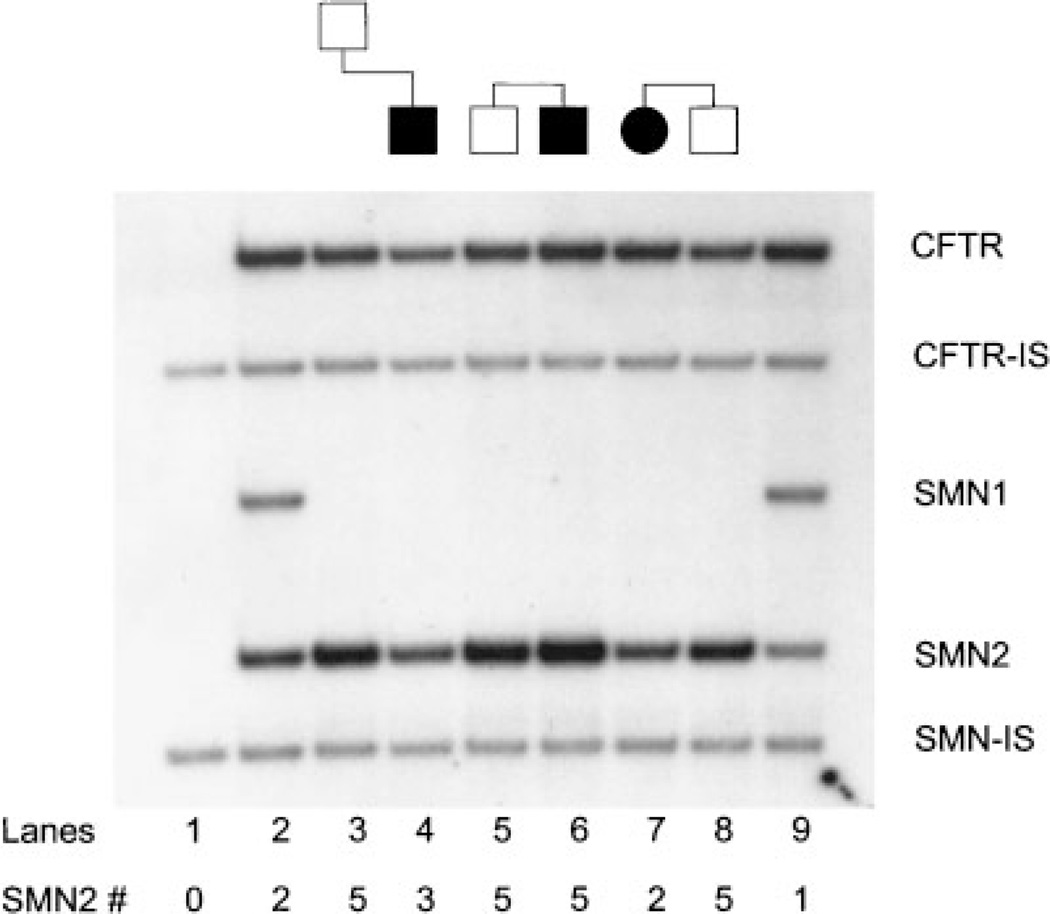
Spinal muscular atrophy is a common autosomal recessive neuromuscular disorder caused by the homozygous loss of the SMN1 gene. The absence of the SMN1 gene has been shown to occur in all types of SMA, childhood and adult forms. In rare cases, asymptomatic family members have also been found with homozygous mutations in the SMN1 gene, suggesting a role for phenotypic modifiers. We describe three unrelated asymptomatic individuals, with family histories of SMA, who were shown to have the homozygous SMN1 deletion. Quantitative studies indicated that the three individuals all had increased SMN2 copy numbers. These cases not only support the role of SMN2 in modifying the phenotype, but our data also demonstrate that expression levels consistent with five copies of the SMN2 genes maybe enough to compensate for the absence of the SMN1 gene. Lastly, in cases similar to the ones described, the measurement of the SMN2 gene copy number may provide valuable prognostic information.
Figures
References
-
- Brahe C, Servidei S, Zappatp S, Ricci E, Tonali P, Neri G. Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet. 1995;346:741–742. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
