

The protein structures that shape caspase activity, specificity, activation and inhibition
- PMID: 15450003
- PMCID: PMC1134104
- DOI: 10.1042/BJ20041142
The protein structures that shape caspase activity, specificity, activation and inhibition
Abstract
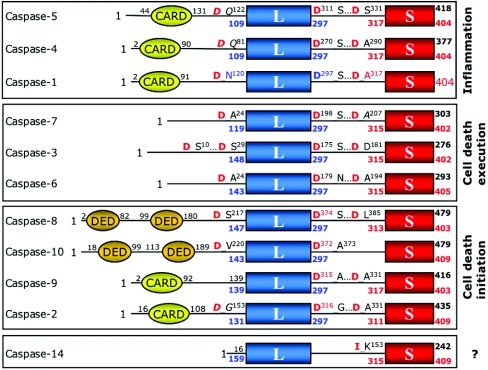
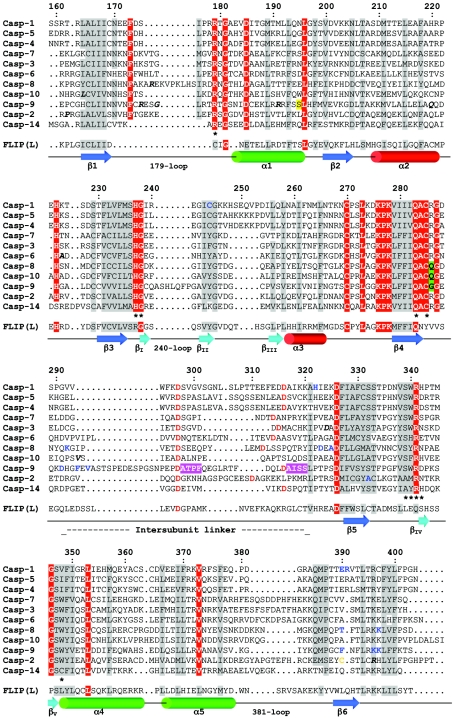
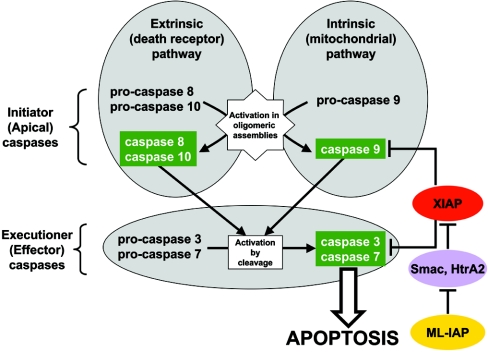
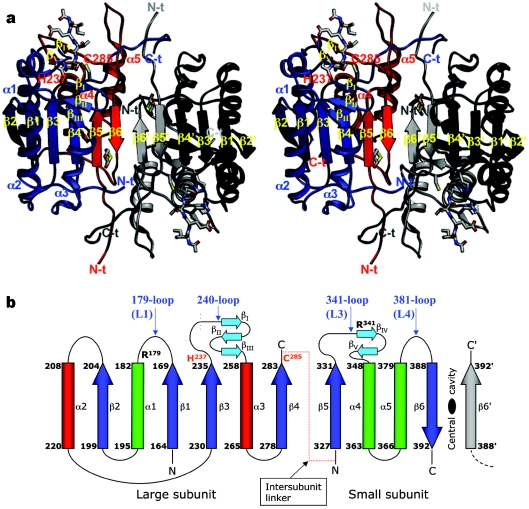
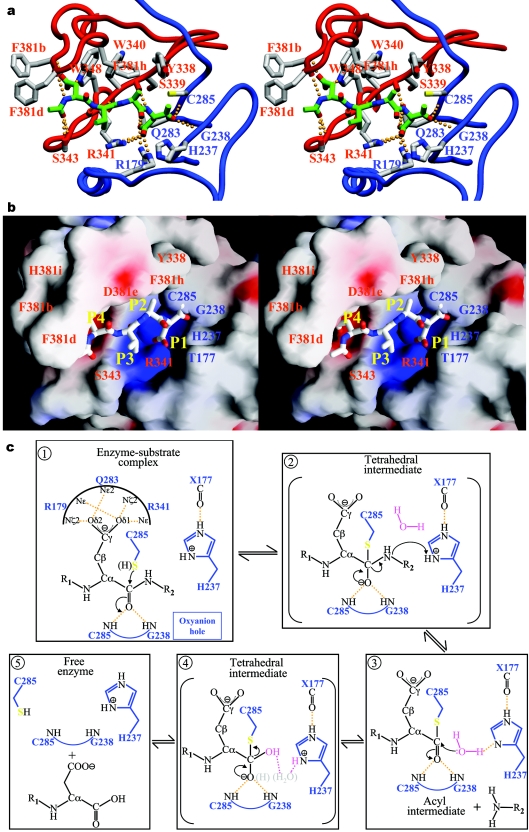
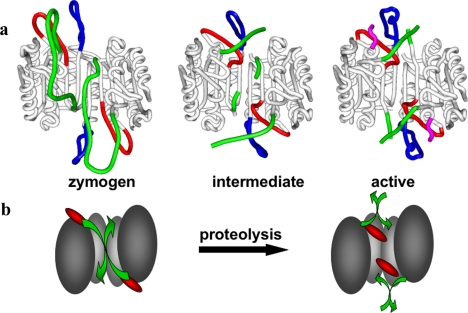
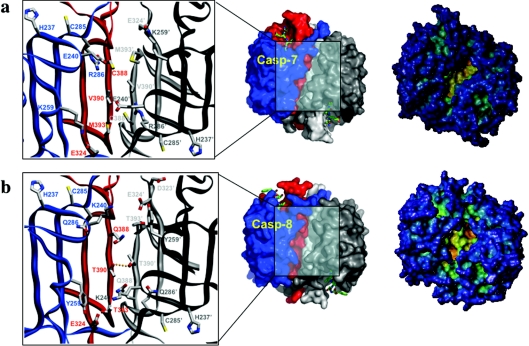
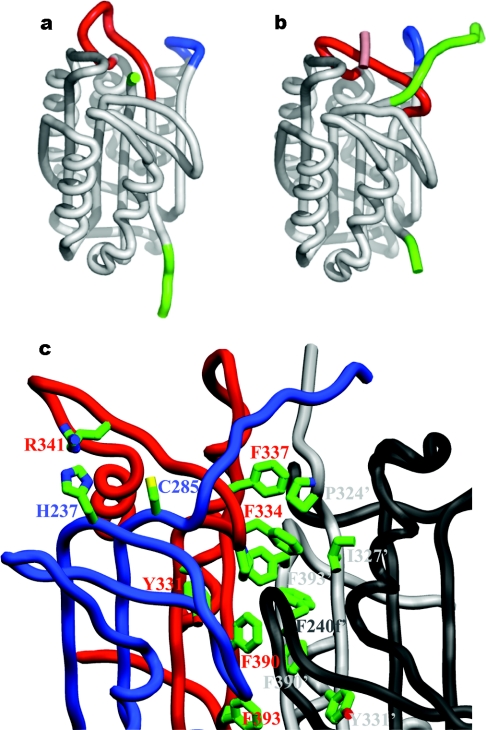
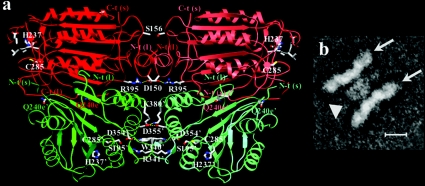
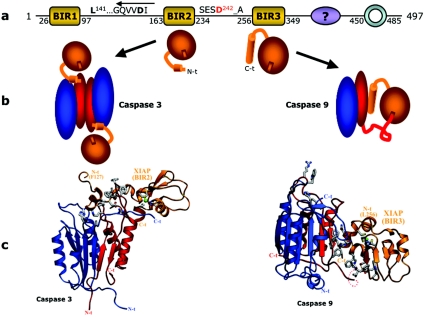
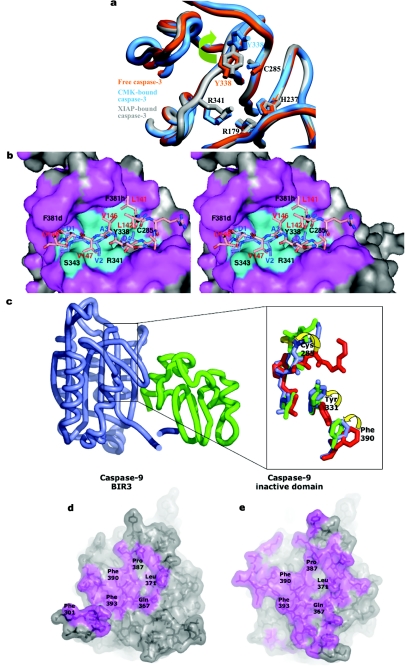
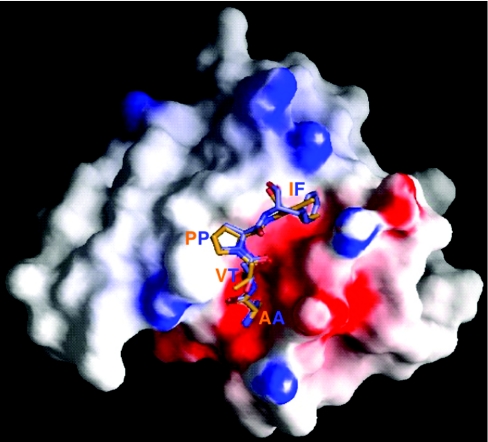
The death morphology commonly known as apoptosis results from a post-translational pathway driven largely by specific limited proteolysis. In the last decade the structural basis for apoptosis regulation has moved from nothing to 'quite good', and we now know the fundamental structures of examples from the initiator phase, the pre-mitochondrial regulator phase, the executioner phase, inhibitors and their antagonists, and even the structures of some substrates. The field is as well advanced as the best known of proteolytic pathways, the coagulation cascade. Fundamentally new mechanisms in protease regulation have been disclosed. Structural evidence suggests that caspases have an unusual catalytic mechanism, and that they are activated by apparently unrelated events, depending on which position in the apoptotic pathway they occupy. Some naturally occurring caspase inhibitors have adopted classic inhibition strategies, but other have revealed completely novel mechanisms. All of the structural and mechanistic information can, and is, being applied to drive therapeutic strategies to combat overactivation of apoptosis in degenerative disease, and underactivation in neoplasia. We present a comprehensive review of the caspases, their regulators and inhibitors from a structural and mechanistic point of view, and with an aim to consolidate the many threads that define the rapid growth of this field.
Figures
References
-
- Schechter I., Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. - PubMed
-
- Thornberry N. A., Bull H. G., Calaycay J. R., Chapman K. T., Howard A. D., Kostura M. J., Miller D. K., Molineaux S. M., Weidner J. R., Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature (London) 1992;356:768–774. - PubMed
-
- Cerretti D. P., Kozlosky C. J., Mosley B., Nelson N., Van Ness K., Greenstreet T. A., March C. J., Kronheim S. R., Druck T., Cannizzaro L. A., et al. Molecular cloning of the interleukin-1β converting enzyme. Science. 1992;256:97–100. - PubMed
-
- Yuan J., Shaham S., Ledoux S., Ellis H. M., Horvitz H. M. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. - PubMed
-
- Alnemri E. S., Livingston D. J., Nicholson D. W., Salvesen G., Thornberry N. A., Wong W. W., Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources