

Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha
- PMID: 15452178
- PMCID: PMC2213285
- DOI: 10.1084/jem.20040634
Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha
Abstract
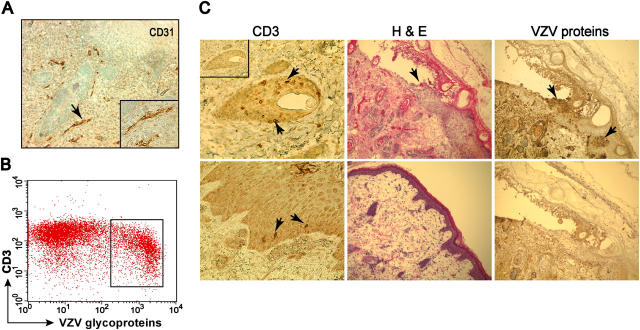
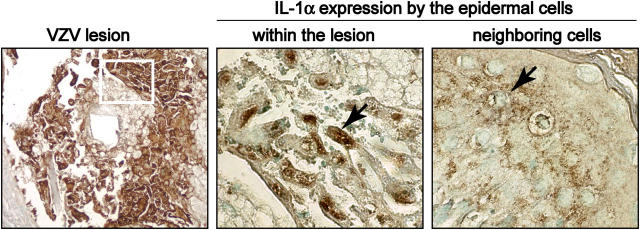
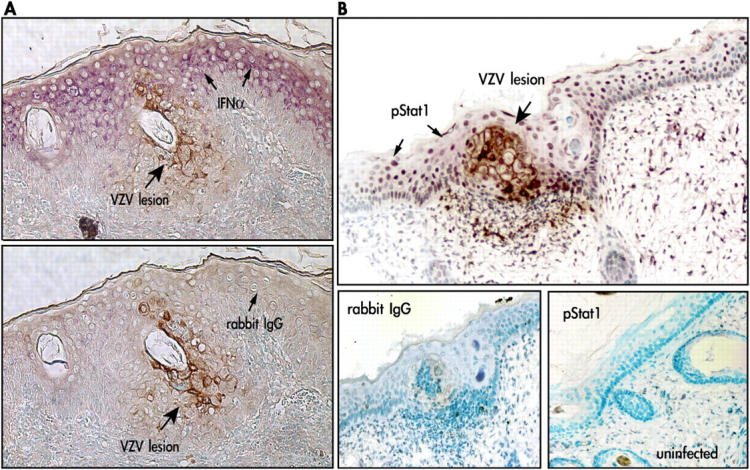
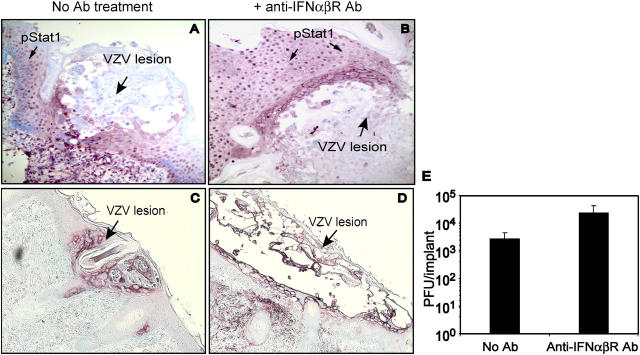
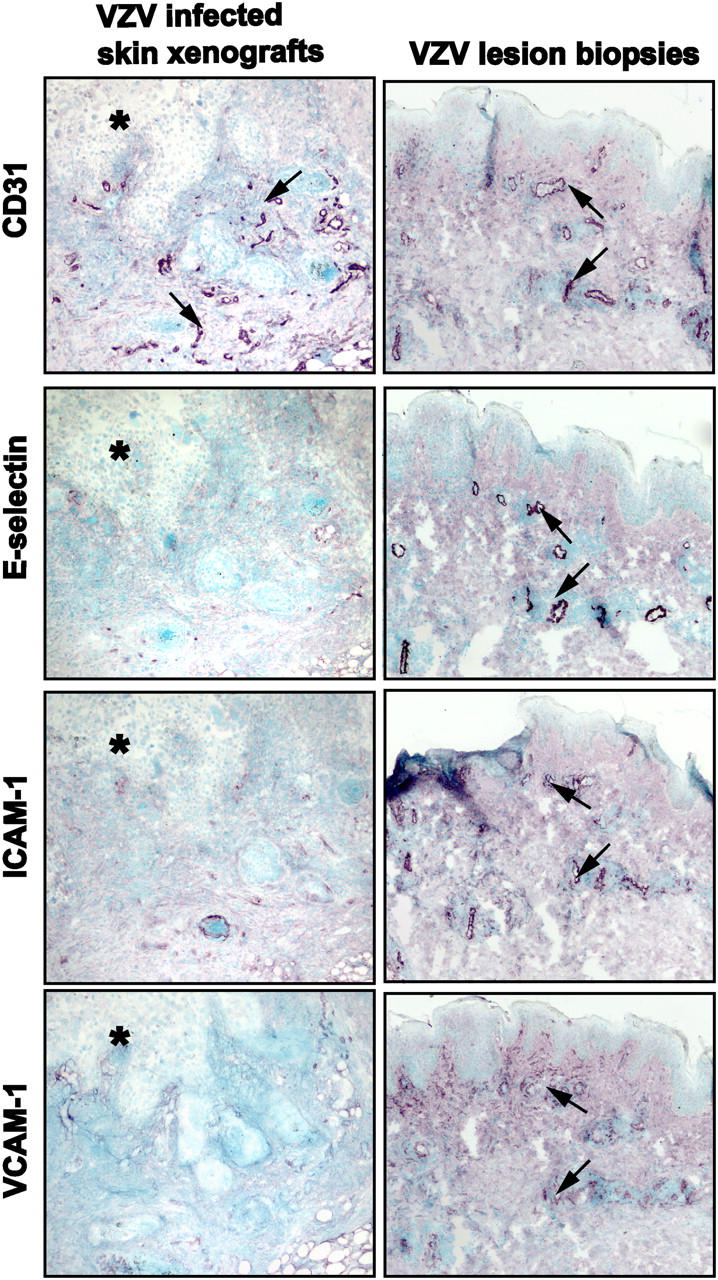
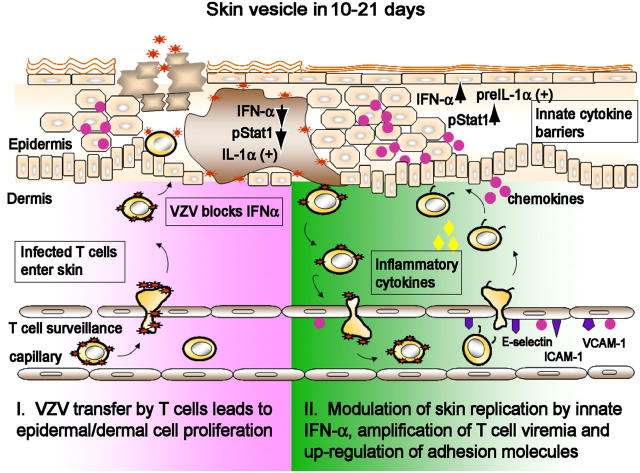
Primary infection with varicella-zoster virus (VZV) causes the characteristic syndrome of varicella, or chickenpox. Experiments in severe combined immunodeficiency mice with human skin grafts (SCIDhu mice) indicate that VZV infection of T cells can mediate transfer of infectious virus to skin. VZV-infected T cells reached epithelial sites of replication within 24 h after entering the circulation. Memory CD4+ T cells were the predominant population recovered from skin in SCIDhu mice given uninfected or infected mononuclear cells, suggesting that immune surveillance by memory T cells may facilitate VZV transfer. The increased susceptibility of memory T cells to VZV infection may further enhance their role in VZV pathogenesis. During VZV skin infection, viral gene products down-regulated interferon-alpha to permit focal replication, whereas adjacent epidermal cells mounted a potent interferon-alpha response against cell-cell spread. Interleukin-1alpha, although activated in VZV-infected cells, did not trigger expression of endothelial adhesion molecules, thereby avoiding early recruitment of inflammatory cells. The prolonged varicella incubation period appears to represent the time required for VZV to overcome antiviral responses of epidermal cells and generate vesicles at the skin surface. Modulation of VZV replication by cutaneous innate immunity may avoid an incapacitating infection of the host that would limit opportunities for VZV transmission.
Figures
Similar articles
-
Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers.J Virol. 2002 Nov;76(22):11425-33. doi: 10.1128/jvi.76.22.11425-11433.2002. J Virol. 2002. PMID: 12388703 Free PMC article.
-
Age-Associated Differences in Infection of Human Skin in the SCID Mouse Model of Varicella-Zoster Virus Pathogenesis.J Virol. 2018 May 14;92(11):e00002-18. doi: 10.1128/JVI.00002-18. Print 2018 Jun 1. J Virol. 2018. PMID: 29563288 Free PMC article.
-
Investigations of the pathogenesis of Varicella zoster virus infection in the SCIDhu mouse model.Herpes. 2006 Nov;13(3):75-80. Herpes. 2006. PMID: 17147912 Review.
-
Varicella-zoster virus infection of a human CD4-positive T-cell line.Virology. 2000 May 10;270(2):278-85. doi: 10.1006/viro.2000.0304. Virology. 2000. PMID: 10792986
-
Varicella-zoster virus T cell tropism and the pathogenesis of skin infection.Curr Top Microbiol Immunol. 2010;342:189-209. doi: 10.1007/82_2010_29. Curr Top Microbiol Immunol. 2010. PMID: 20397071 Free PMC article. Review.
Cited by
-
Stroke-like Symptoms as Presenting Signs of Varicella Zoster Meningitis in an Immunocompetent Adult.Cureus. 2022 Feb 9;14(2):e22062. doi: 10.7759/cureus.22062. eCollection 2022 Feb. Cureus. 2022. PMID: 35295357 Free PMC article.
-
Herpes Zoster in Patients Receiving JAK Inhibitors For Ulcerative Colitis: Mechanism, Epidemiology, Management, and Prevention.Inflamm Bowel Dis. 2018 Sep 15;24(10):2173-2182. doi: 10.1093/ibd/izy150. Inflamm Bowel Dis. 2018. PMID: 29788127 Free PMC article. Review.
-
Varicella Zoster Virus infects mucosal associated Invariant T cells.Front Immunol. 2023 Mar 17;14:1121714. doi: 10.3389/fimmu.2023.1121714. eCollection 2023. Front Immunol. 2023. PMID: 37006246 Free PMC article.
-
Creating the "Dew Drop on a Rose Petal": the Molecular Pathogenesis of Varicella-Zoster Virus Skin Lesions.Microbiol Mol Biol Rev. 2023 Sep 26;87(3):e0011622. doi: 10.1128/mmbr.00116-22. Epub 2023 Jun 24. Microbiol Mol Biol Rev. 2023. PMID: 37354037 Free PMC article. Review.
-
Immunogenicity and cellular response of a herpes zoster virus gEgI fusion protein adjuvanted with CpG-emulsion in mice.J Nanobiotechnology. 2025 May 30;23(1):395. doi: 10.1186/s12951-025-03423-w. J Nanobiotechnology. 2025. PMID: 40448056 Free PMC article.
References
-
- Arvin, A. 2001. Chapter 79. Varicella-Zoster Virus. Fields Virology. D. Knipe and P. Howley, editors. Lippincott Williams & Wilkins, Philadelphia, PA. 2731–2767.
-
- Grose, C. 1981. Variation on a theme by Fenner: the pathogenesis of chickenpox. Pediatrics. 68:735–737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
