

A model of the putative pore region of the cardiac ryanodine receptor channel
- PMID: 15454434
- PMCID: PMC1304657
- DOI: 10.1529/biophysj.104.044180
A model of the putative pore region of the cardiac ryanodine receptor channel
Abstract
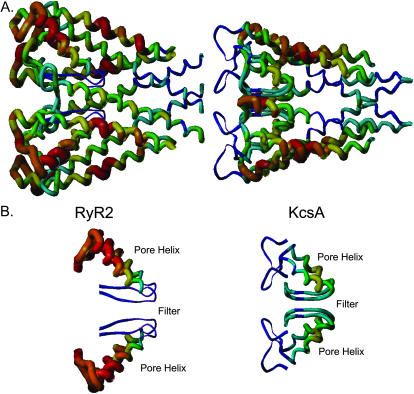
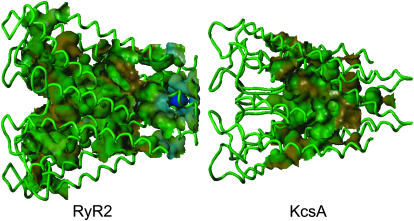
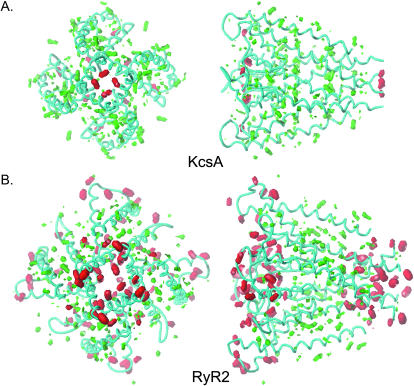
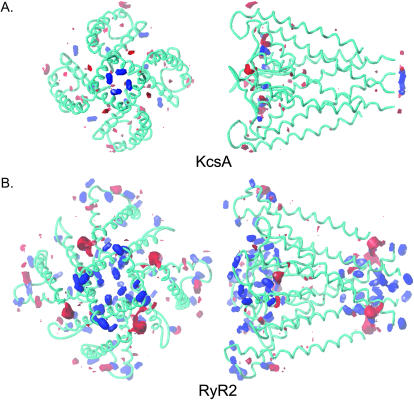
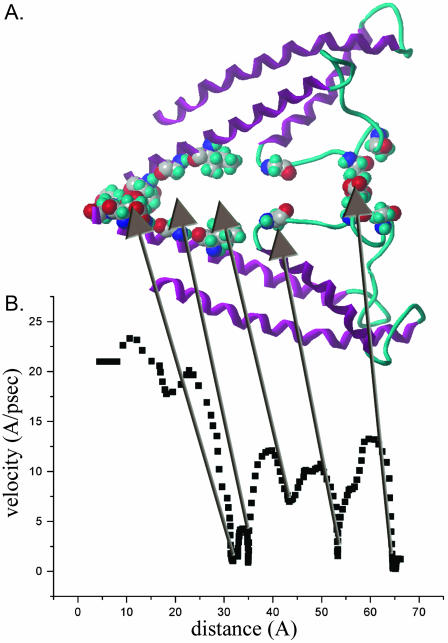
Using the bacterial K+ channel KcsA as a template, we constructed models of the pore region of the cardiac ryanodine receptor channel (RyR2) monomer and tetramer. Physicochemical characteristics of the RyR2 model monomer were compared with the template, including homology, predicted secondary structure, surface area, hydrophobicity, and electrostatic potential. Values were comparable with those of KcsA. Monomers of the RyR2 model were minimized and assembled into a tetramer that was, in turn, minimized. The assembled tetramer adopts a structure equivalent to that of KcsA with a central pore. Characteristics of the RyR2 model tetramer were compared with the KcsA template, including average empirical energy, strain energy, solvation free energy, solvent accessibility, and hydrophobic, polar, acid, and base moments. Again, values for the model and template were comparable. The pores of KcsA and RyR2 have a common motif with a hydrophobic channel that becomes polar at both entrances. Quantitative comparisons indicate that the assembled structure provides a plausible model for the pore of RyR2. Movement of Ca2+, K+, and tetraethylammonium (TEA+) through the model RyR2 pore were simulated with explicit solvation. These simulations suggest that the model RyR2 pore is permeable to Ca2+ and K+ with rates of translocation greater for K+. In contrast, simulations indicate that tetraethylammonium blocks movement of metal cations.
Copyright 2004 Biophysical Society
Figures






References
-
- Anyatonwu, G. I., E. D. Buck, and B. E. Ehrlich. 2003. Methanethiosulfonate ethylammonium block of amine currents through the ryanodine receptor reveals single pore architecture. J. Biol. Chem. 278:45528–45538. - PubMed
-
- Berridge, M. J., M. D. Bootman, and H. L. Roderick. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517–529. - PubMed
-
- Dayhoff, M. O., R. M. Schwartz, and B. C. Orcutt. 1978. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure. M. O. Dayhoff, editor. National Biomedical Research Foundation, Silver Spring, MD. 345–52.
-
- Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. L. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
