

Soft docking and multiple receptor conformations in virtual screening
- PMID: 15456251
- PMCID: PMC1413506
- DOI: 10.1021/jm049756p
Soft docking and multiple receptor conformations in virtual screening
Abstract
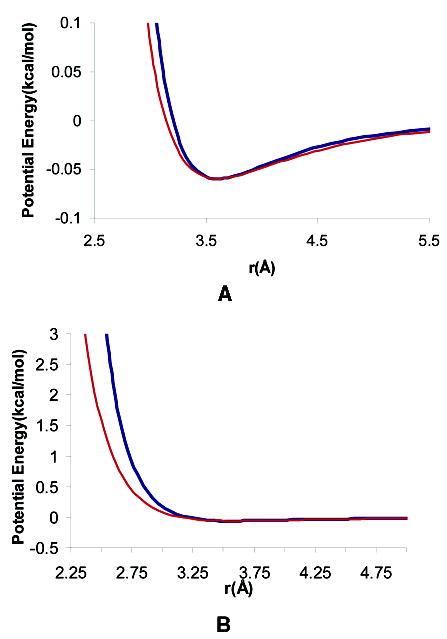
Protein conformational change is an important consideration in ligand-docking screens, but it is difficult to predict. A simple way to account for protein flexibility is to soften the criterion for steric fit between ligand and receptor. A more comprehensive but more expensive method would be to sample multiple receptor conformations explicitly. Here, these two approaches are compared. A "soft" scoring function was created by attenuating the repulsive term in the Lennard-Jones potential, allowing for a closer approach between ligand and protein. The standard, "hard" Lennard-Jones potential was used for docking to multiple receptor conformations. The Available Chemicals Directory (ACD) was screened against two cavity sites in the T4 lysozyme. These sites undergo small but significant conformational changes on ligand binding, making them good systems for soft docking. The ACD was also screened against the drug target aldose reductase, which can undergo large conformational changes on ligand binding. We evaluated the ability of the scoring functions to identify known ligands from among the over 200 000 decoy molecules in the database. The soft potential was always better at identifying known ligands than the hard scoring function when only a single receptor conformation was used. Conversely, the soft function was worse at identifying known leads than the hard function when multiple receptor conformations were used. This was true even for the cavity sites and was especially true for aldose reductase. To test the multiple-conformation method predictively, we screened the ACD for molecules that preferentially docked to the expanded conformation of aldose reductase, known to bind larger ligands. Six novel molecules that ranked among the top 0.66% of hits from the multiple-conformation calculation, but ranked relatively poorly in the soft docking calculation, were tested experimentally for enzyme inhibition. Four of these six inhibited the enzyme, the best with an IC(50) of 8 microM. Although ligands can get better scores in soft docking, the same is also true for decoys. The improved ranking of such decoys can come at the expense of true ligands.
Figures






References
-
- Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997;Suppl:215–220. - PubMed
-
- Schnecke V, Swanson CA, Getzoff ED, Tainer JA, Kuhn LA. Screening a peptidyl database for potential ligands to proteins with side-chain flexibility. Proteins. 1998;33:74–87. - PubMed
-
- Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. - PubMed
-
- Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994;235:345–356. - PubMed
-
- Kallblad P, Dean PM. Efficient conformational sampling of local side-chain flexibility. J Mol Biol. 2003;326:1651–1665. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Chemical Information
