

Glycogen synthase kinase 3beta is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase
- PMID: 15466414
- PMCID: PMC5328675
- DOI: 10.1074/jbc.M408607200
Glycogen synthase kinase 3beta is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase
Abstract
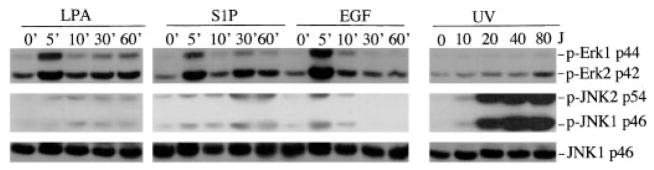
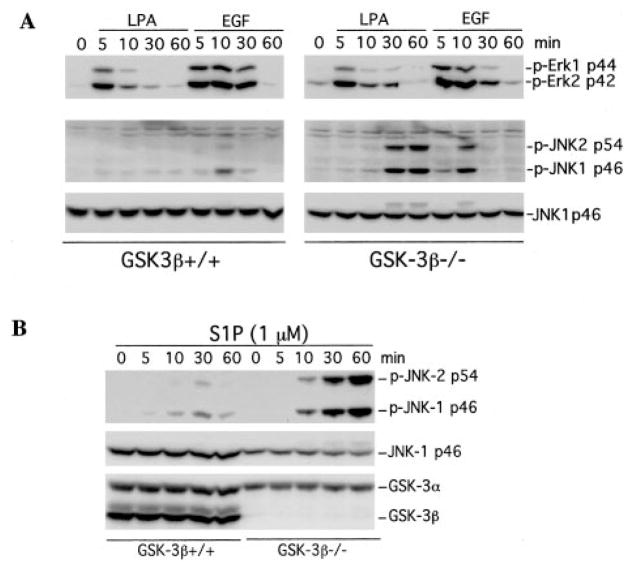
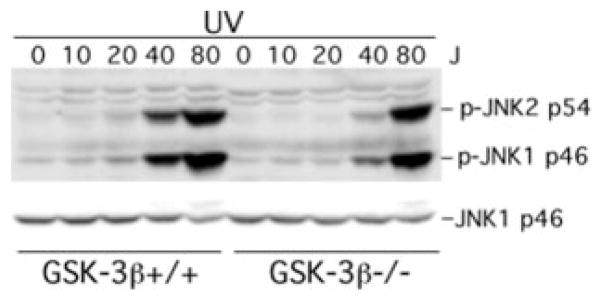
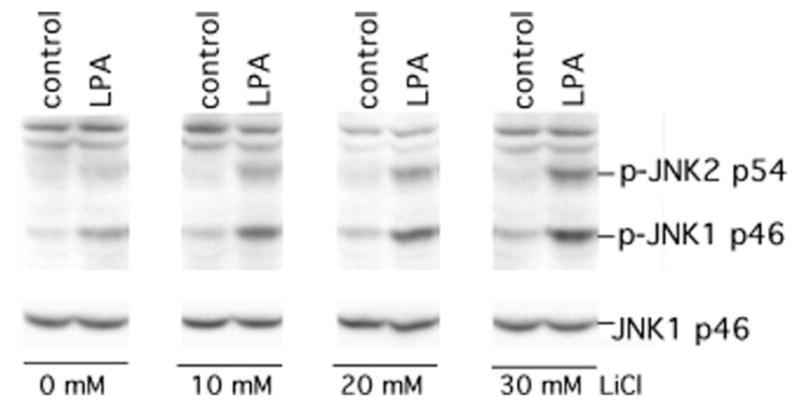
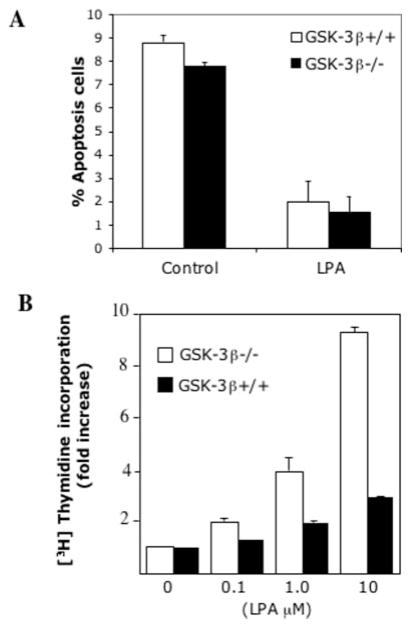
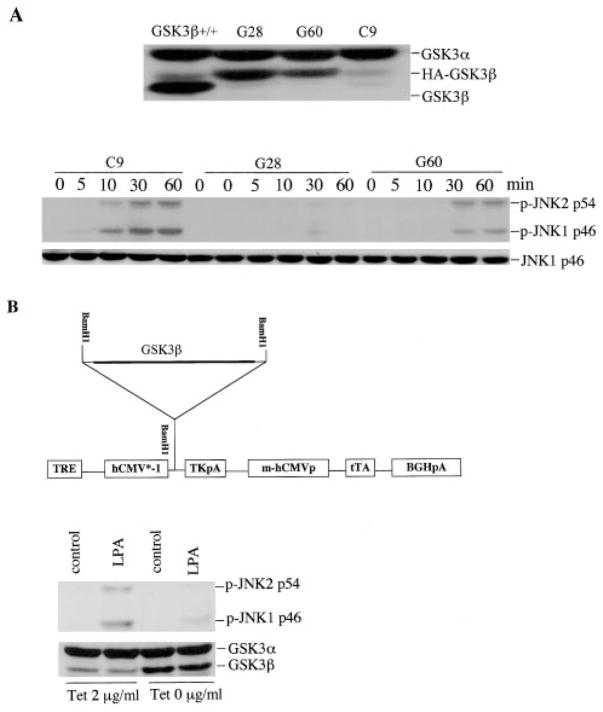
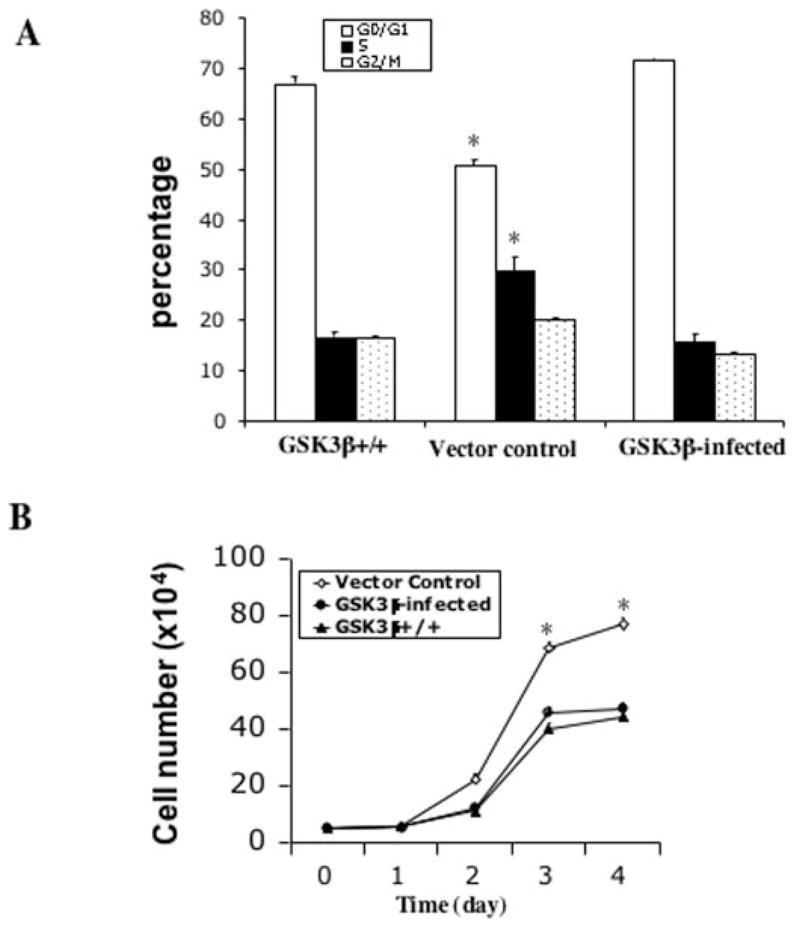
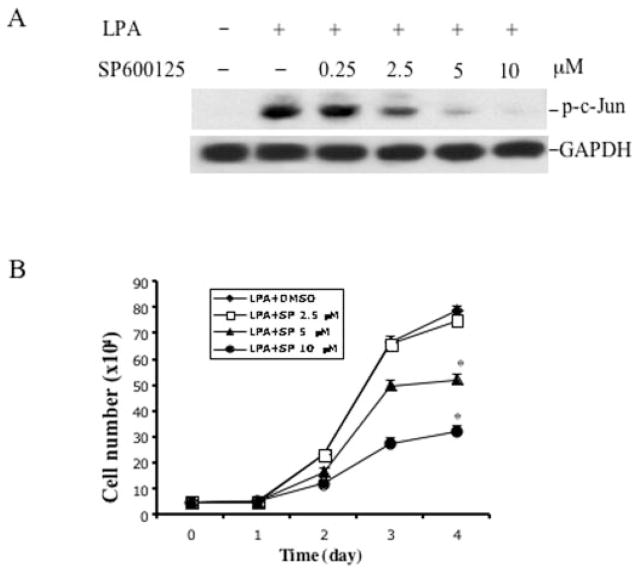
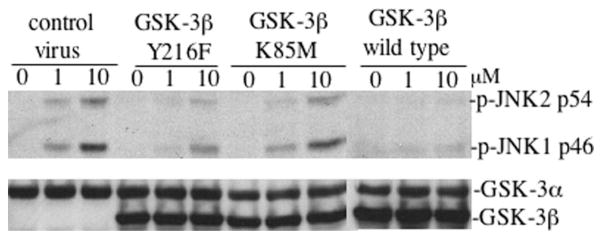
The c-Jun N-terminal kinase (JNK)/stress activated protein kinase is preferentially activated by stress stimuli. Growth factors, particularly ligands for G protein-coupled receptors, usually induce only modest JNK activation, although they may trigger marked activation of the related extracellular signal-regulated kinase. In the present study, we demonstrated that homozygous disruption of glycogen synthase kinase 3beta (GSK-3beta) dramatically sensitized mouse embryonic fibroblasts (MEFs) to JNK activation induced by lysophosphatidic acid (LPA) and sphingosine-1-phosphate, two prototype ligands for G protein-coupled receptors. To a lesser degree, a lack of GSK-3beta also potentiated JNK activation in response to epidermal growth factor. In contrast, the absence of GSK-3beta decreased UV light-induced JNK activation. The increased JNK activation induced by LPA in GSK-3beta null MEFs was insufficient to trigger apoptotic cell death or growth inhibition. Instead, the increased JNK activation observed in GSK-3beta-/- MEFs was associated with an increased proliferative response to LPA, which was reduced by the inhibition of JNK. Ectopic expression of GSK-3beta in GSK-3beta-negative MEFs restrained LPA-triggered JNK phosphorylation and induced a concomitant decrease in the mitogenic response to LPA compatible with GSK-3beta through the inhibition of JNK activation, thus limiting LPA-induced cell proliferation. Mutation analysis indicated that GSK-3beta kinase activity was required for GSK-3beta to optimally inhibit LPA-stimulated JNK activation. Thus GSK-3beta serves as a physiological switch to specifically repress JNK activation in response to LPA, sphingosine-1-phosphate, or the epidermal growth factor. These results reveal a novel role for GSK-3beta in signal transduction and cellular responses to growth factors.
Figures
References
-
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
