

A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration
- PMID: 15483130
- PMCID: PMC6730049
- DOI: 10.1523/JNEUROSCI.2729-04.2004
A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration
Abstract
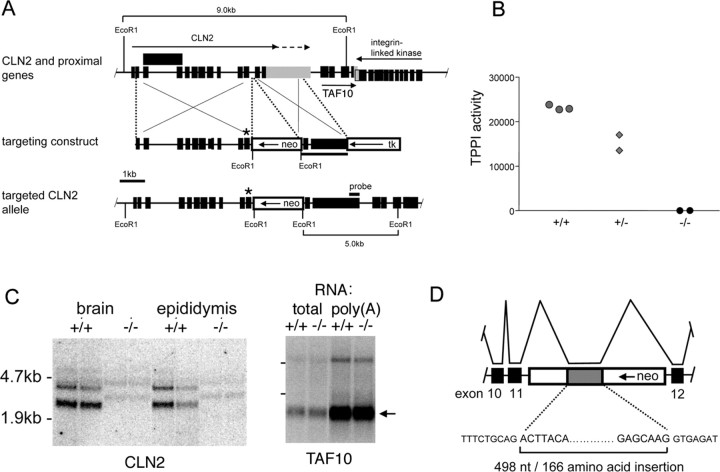
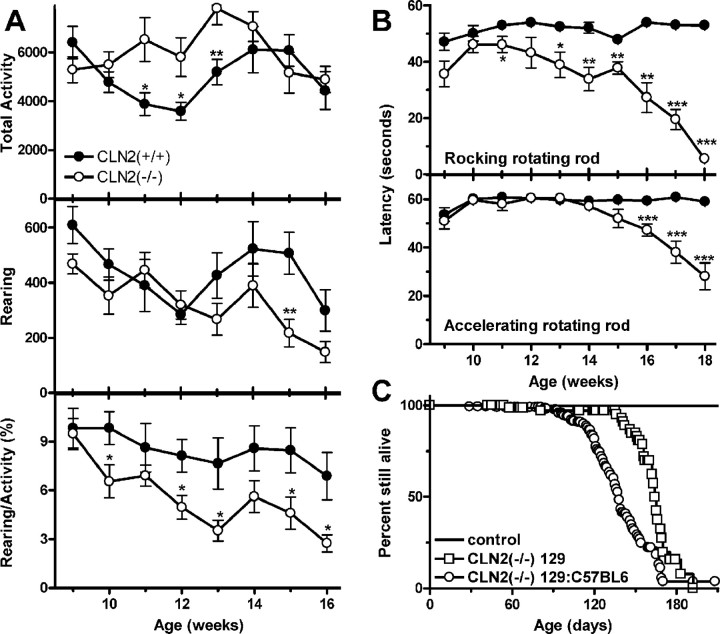
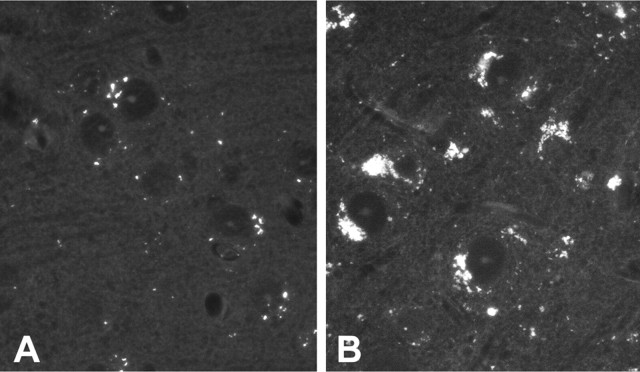
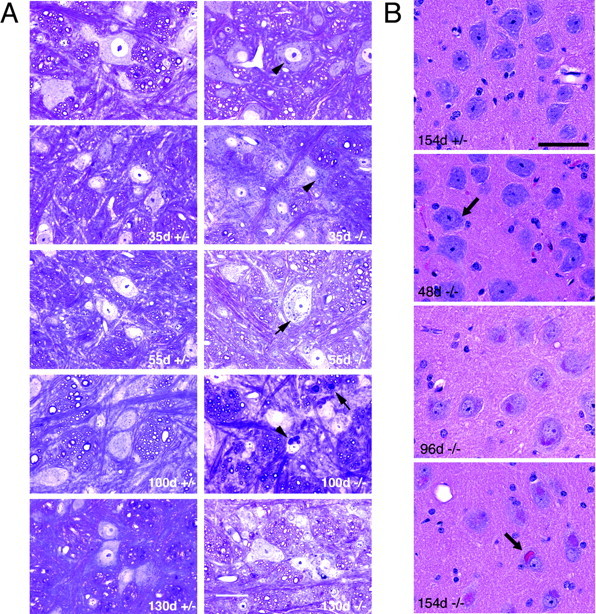
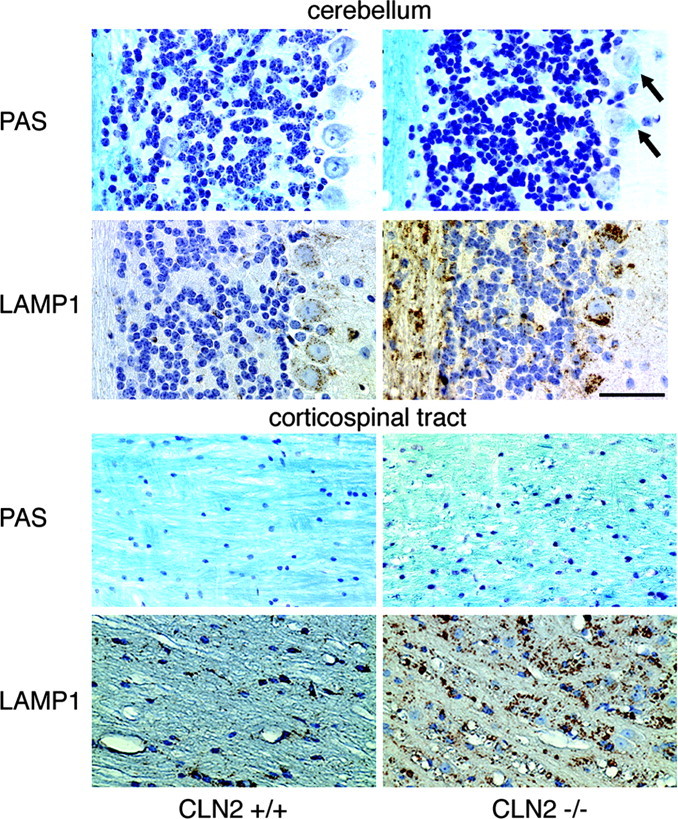
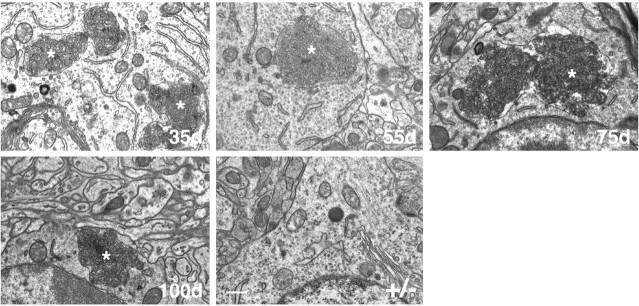
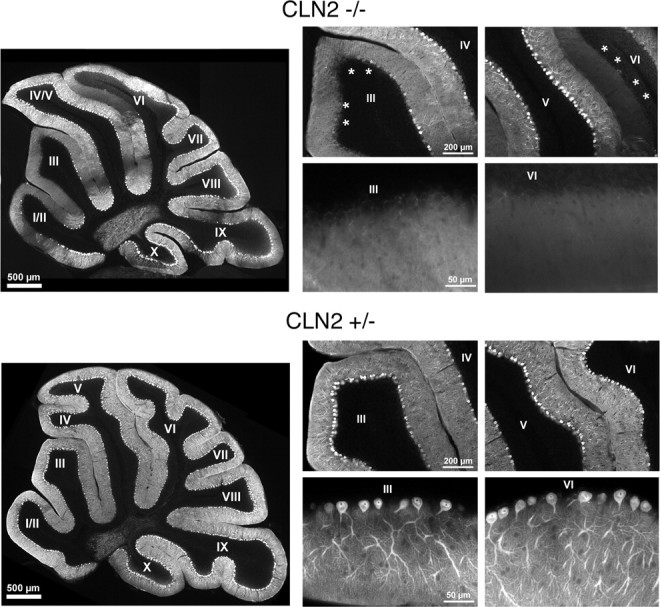
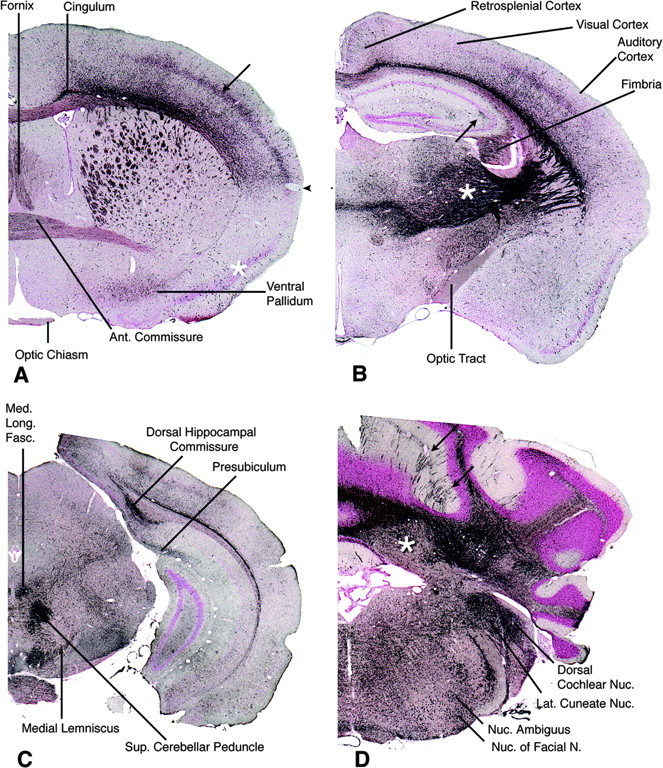
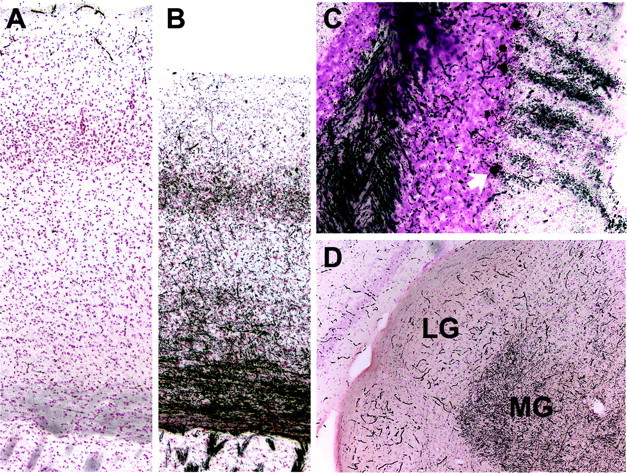
Mutations in the CLN2 gene, which encodes a lysosomal serine protease, tripeptidyl-peptidase I (TPP I), result in an autosomal recessive neurodegenerative disease of children, classical late-infantile neuronal ceroid lipofuscinosis (cLINCL). cLINCL is inevitably fatal, and there currently exists no cure or effective treatment. In this report, we provide the characterization of the first CLN2-targeted mouse model for cLINCL. CLN2-targeted mice were fertile and apparently healthy at birth despite an absence of detectable TPP I activity. At approximately 7 weeks of age, neurological deficiencies became evident with the onset of a tremor that became progressively more severe and was eventually accompanied by ataxia. Lifespan of the affected mice was greatly reduced (median survival, 138 d), and extensive neuronal pathology was observed including a prominent accumulation of cytoplasmic storage material within the lysosomal-endosomal compartment, a loss of cerebellar Purkinje cells, and widespread axonal degeneration. The CLN2-targeted mouse therefore recapitulates much of the pathology and clinical features of cLINCL and represents an animal model that should provide clues to the normal cellular function of TPP I and the pathogenic processes that underlie neuronal death in its absence. In addition, the CLN2-targeted mouse also represents a valuable model for the evaluation of different therapeutic strategies.
Figures
References
-
- Bronson RT, Donahue LR, Johnson KR, Tanner A, Lane PW, Faust JR (1998) Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am J Med Genet 77: 289-297. - PubMed
-
- Chen JW, Pan W, D'Souza MP, August JT (1985) Lysosome-associated membrane proteins: characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch Biochem Biophys 239: 574-586. - PubMed
-
- DeOlmos JS, Ingram WR (1971) An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res 33: 523-529. - PubMed
-
- Elleder M, Franc J, Kraus J, Nevsimalova S, Sixtova K, Zeman J (1997) Neuronal ceroid lipofuscinosis in the Czech Republic: analysis of 57 cases. Report of the `Prague NCL group'. Eur J Paediatr Neurol 1: 109-114. - PubMed
-
- Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME (2002) Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet 70: 324-335. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials