

Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation
- PMID: 15492925
- PMCID: PMC1182143
- DOI: 10.1086/426462
Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation
Abstract
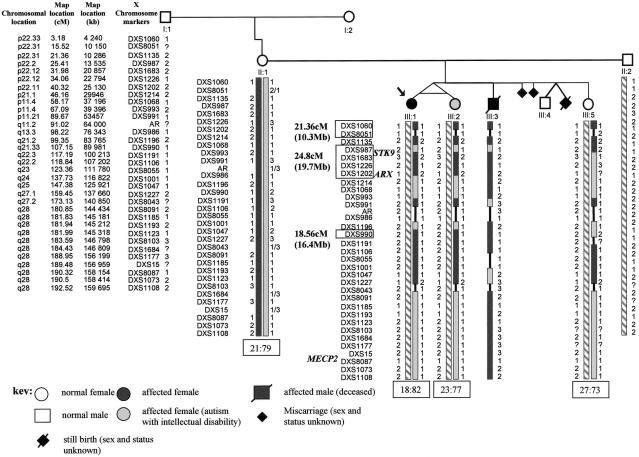
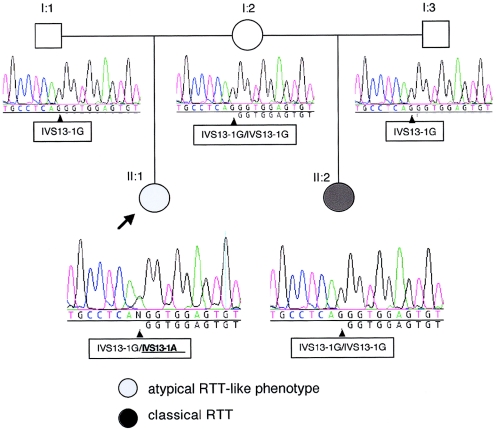
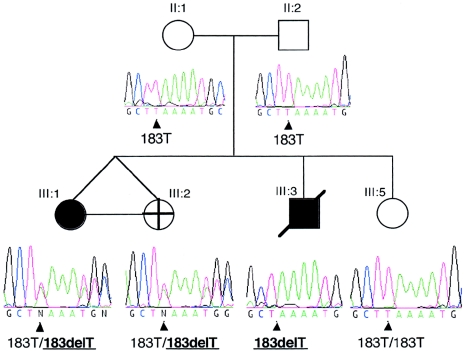
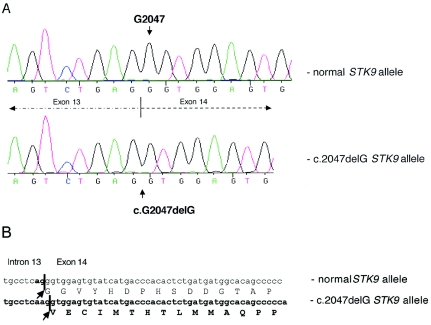
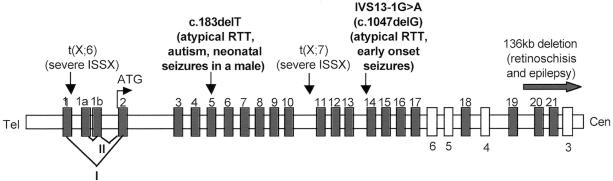
Rett syndrome (RTT) is a severe neurodevelopmental disorder caused, in most classic cases, by mutations in the X-linked methyl-CpG-binding protein 2 gene (MECP2). A large degree of phenotypic variation has been observed in patients with RTT, both those with and without MECP2 mutations. We describe a family consisting of a proband with a phenotype that showed considerable overlap with that of RTT, her identical twin sister with autistic disorder and mild-to-moderate intellectual disability, and a brother with profound intellectual disability and seizures. No pathogenic MECP2 mutations were found in this family, and the Xq28 region that contains the MECP2 gene was not shared by the affected siblings. Three other candidate regions were identified by microsatellite mapping, including 10.3 Mb at Xp22.31-pter between Xpter and DXS1135, 19.7 Mb at Xp22.12-p22.11 between DXS1135 and DXS1214, and 16.4 Mb at Xq21.33 between DXS1196 and DXS1191. The ARX and CDKL5 genes, both of which are located within the Xp22 region, were sequenced in the affected family members, and a deletion of nucleotide 183 of the coding sequence (c.183delT) was identified in CDKL5 in the affected family members. In a screen of 44 RTT cases, a single splice-site mutation, IVS13-1G-->A, was identified in a girl with a severe phenotype overlapping RTT. In the mouse brain, Cdkl5 expression overlaps--but is not identical to--that of Mecp2, and its expression is unaffected by the loss of Mecp2. These findings confirm CDKL5 as another locus associated with epilepsy and X-linked mental retardation. These results also suggest that mutations in CDKL5 can lead to a clinical phenotype that overlaps RTT. However, it remains to be determined whether CDKL5 mutations are more prevalent in specific clinical subgroups of RTT or in other clinical presentations.
Figures
References
Electronic-Database Information
-
- Cedar Genetics Map, http://cedar.genetics.soton.ac.uk/pub/chromX/gmap (for map of the X chromosome and genetic map distances)
-
- Ensembl Human Genome Browser, http://www.ensembl.org/Homo_sapiens/mapview?chr=X (for MapView and genetic map distances)
-
- Genome Database, http://gdbwww.gdb.org/ (for microsatellite information)
-
- Integrated X Chromosome Database, version 2.3, http://ixdb.molgen.mpg.de/ (for genetic map distances)
-
- Mouse Genome Informatics (MGI), http://www.informatics.jax.org/ (for details of the mouse Cdkl5 sequence)
References
-
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, Lichtarge O, Smith EO, Glaze DG, Zoghbi HY (2000) Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol 47:670–67910.1002/1531-8249(200005)47:5<670::AID-ANA20>3.3.CO;2-6 - DOI - PubMed
-
- Ben Zeev B, Yaron Y, Schanen NC, Wolf H, Brandt N, Ginot N, Shomrat R, Orr-Urtreger A (2002) Rett syndrome: clinical manifestations in males with MECP2 mutations. J Child Neurol 17:20–24 - PubMed
-
- Bienvenu T, Poirer K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet MC, Francis F, Couvert P, Gomot M, Moraine C, Van Bokhoven H, Kalscheuer V, Frints S, Gecz F, Ohzaki K, Chaabouni H, Fryns JP, Desportes V, Beldjord C, Chelly J (2002) ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 11:981–99110.1093/hmg/11.8.981 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
