

The cytoskeleton in neurodegenerative diseases
- PMID: 15495240
- PMCID: PMC3011821
- DOI: 10.1002/path.1650
The cytoskeleton in neurodegenerative diseases
Abstract
Abundant abnormal aggregates of cytoskeletal proteins are neuropathological signatures of many neurodegenerative diseases that are broadly classified by filamentous aggregates of neuronal intermediate filament (IF) proteins, or by inclusions containing the microtubule-associated protein (MAP) tau. The discovery of mutations in neuronal IF and tau genes firmly establishes the importance of neuronal IF proteins and tau in the pathogenesis of neurodegenerative diseases. Multiple IF gene mutations are pathogenic for Charcot-Marie-Tooth (CMT) disease and amyotrophic lateral sclerosis (ALS)--in addition to those in the copper/zinc superoxide dismutase-1 (SOD1) gene. Tau gene mutations are pathogenic for frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), and tau polymorphisms are genetic risk factors for sporadic progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). Thus, IF and tau abnormalities are linked directly to the aetiology and pathogenesis of neurodegenerative diseases. In vitro and transgenic animal models are being used to demonstrate that different mutations impair protein function, promote tau fibrilization, or perturb tau gene splicing, leading to aberrant and distinct tau aggregates. For recognition of these disorders at neuropathological examination, immunohistochemistry is needed, and this may be combined with biochemistry and molecular genetics to properly determine the nosology of a particular case. As reviewed here, the identification of molecular genetic defects and biochemical alterations in cytoskeletal proteins of human neurodegenerative diseases has facilitated experimental studies and will promote the development of assays of molecules which inhibit abnormal neuronal IF and tau protein inclusions.
Copyright (c) 2004 Pathological Society of Great Britain and Ireland.
Figures

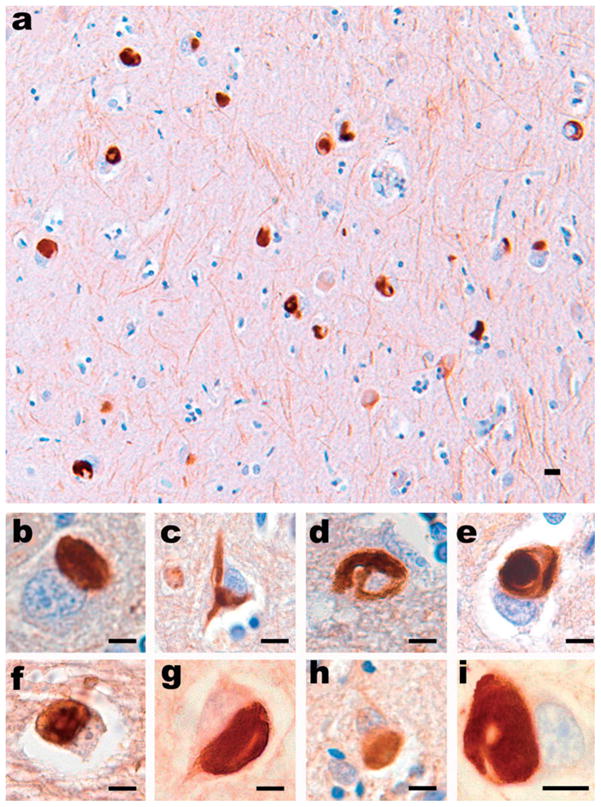
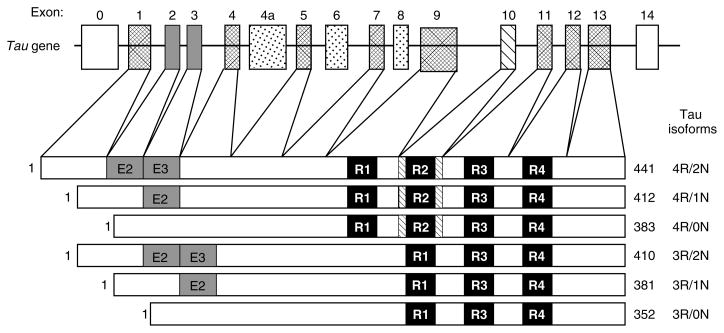
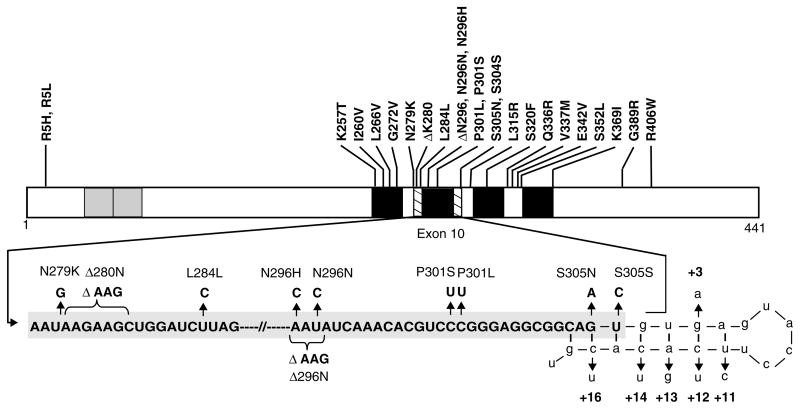
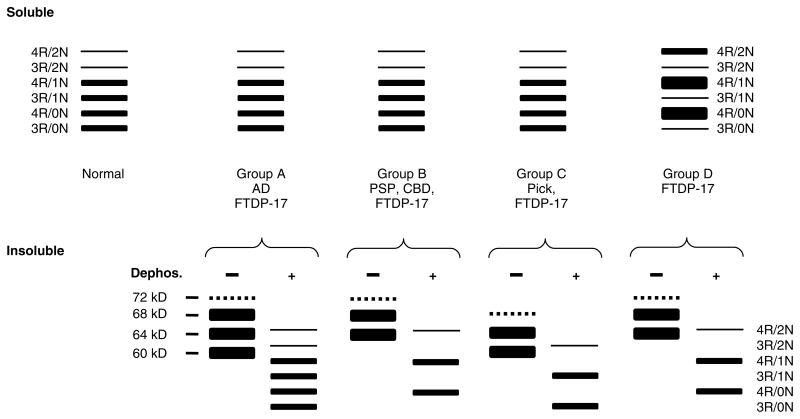
References
-
- Lee K, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci. 1996;19:187–217. - PubMed
-
- Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. - PubMed
-
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:55–56. - PubMed
-
- Al-Chalabi A, Miller CCJ. Neurofilaments and neurological disease. Bioessays. 2003;25:346–365. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
