

To be folded or to be unfolded?
- PMID: 15498936
- PMCID: PMC2286584
- DOI: 10.1110/ps.04881304
To be folded or to be unfolded?
Abstract
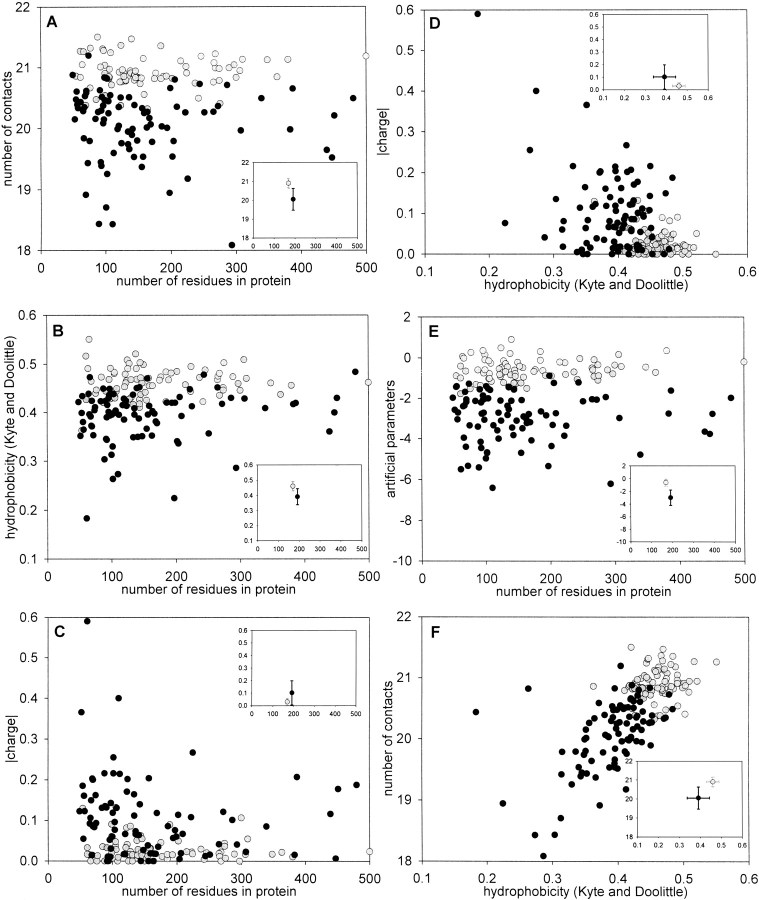
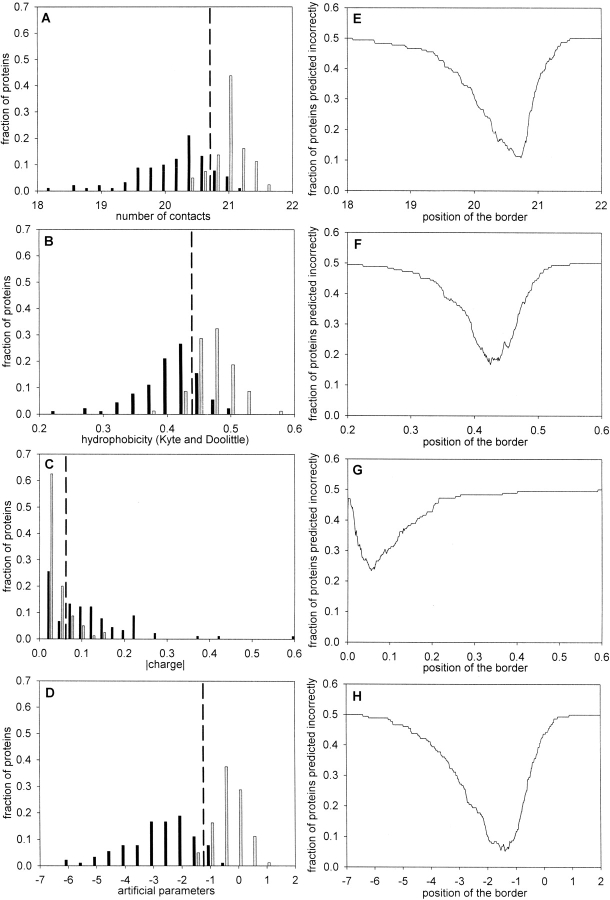
The lack of ordered structure in "natively unfolded" proteins raises a general question: Are there intrinsic properties of amino acid residues that are responsible for the absence of fixed structure at physiological conditions? In this article, we demonstrate that the competence of a protein to be folded or to be unfolded may be determined by the property of amino acid residues to form a sufficient number of contacts in a globular state. The expected average number of contacts per residue calculated from the amino acid sequence alone (using the average number of contacts for 20 amino acid residues in globular proteins) can be used as one of the simple indicators of natively unfolded proteins. The prediction accuracy for the sets of 80 folded and 90 natively unfolded proteins reaches 89% if the expected average number of contacts is used as a parameter and 83% in the case of hydrophobicity. An optimal set of artificial parameters for 20 amino acid residues obtained by Monte Carlo algorithm to maximally separate the sets of 90 natively unfolded and 80 folded proteins demonstrates the upper limit for prediction accuracy, which is 95%.
Figures
Similar articles
-
[Prediction of natively unfolded regions in protein chain].Mol Biol (Mosk). 2006 Mar-Apr;40(2):341-8. Mol Biol (Mosk). 2006. PMID: 16637275 Russian.
-
Native protein sequences are designed to destabilize folding intermediates.Biochemistry. 2006 Feb 28;45(8):2488-92. doi: 10.1021/bi0523714. Biochemistry. 2006. PMID: 16489741
-
Folding protein alpha-carbon chains into compact forms by Monte Carlo methods.Proteins. 1992 Nov;14(3):409-20. doi: 10.1002/prot.340140310. Proteins. 1992. PMID: 1438179 Review.
-
On the NMR analysis of pKa values in the unfolded state of proteins by extrapolation to zero denaturant.Biophys Chem. 2007 Sep;129(2-3):242-50. doi: 10.1016/j.bpc.2007.06.004. Epub 2007 Jun 14. Biophys Chem. 2007. PMID: 17611012
-
NMR characterization of partially folded and unfolded conformational ensembles of proteins.Biopolymers. 1999;51(3):191-207. doi: 10.1002/(SICI)1097-0282(1999)51:3<191::AID-BIP3>3.0.CO;2-B. Biopolymers. 1999. PMID: 10516571 Review.
Cited by
-
Predicting mostly disordered proteins by using structure-unknown protein data.BMC Bioinformatics. 2007 Mar 6;8:78. doi: 10.1186/1471-2105-8-78. BMC Bioinformatics. 2007. PMID: 17338828 Free PMC article.
-
Large-scale prediction of long disordered regions in proteins using random forests.BMC Bioinformatics. 2009 Jan 7;10:8. doi: 10.1186/1471-2105-10-8. BMC Bioinformatics. 2009. PMID: 19128505 Free PMC article.
-
Prediction of amyloidogenic and disordered regions in protein chains.PLoS Comput Biol. 2006 Dec 29;2(12):e177. doi: 10.1371/journal.pcbi.0020177. Epub 2006 Nov 6. PLoS Comput Biol. 2006. PMID: 17196033 Free PMC article.
-
A comprehensive overview of computational protein disorder prediction methods.Mol Biosyst. 2012 Jan;8(1):114-21. doi: 10.1039/c1mb05207a. Epub 2011 Aug 26. Mol Biosyst. 2012. PMID: 21874190 Free PMC article. Review.
-
Fast and accurate multivariate Gaussian modeling of protein families: predicting residue contacts and protein-interaction partners.PLoS One. 2014 Mar 24;9(3):e92721. doi: 10.1371/journal.pone.0092721. eCollection 2014. PLoS One. 2014. PMID: 24663061 Free PMC article.
References
-
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer, E.F., Brice, M.D., Rogers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur. J. Biochem. 80 319–324. - PubMed
-
- Dunker, A.K., Garner, E., Guilliot, S., Romero, P., Albrecht, K., Hart, J., Obradovic, Z., Kissinger, C., and Villafranca, J.E. 1998. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 473–484. - PubMed
-
- Dyson, H.J. and Wright, P.E. 2002. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 62 311–340. - PubMed
-
- Fauchere, I.I. and Pliska, V. 1983. Hydrophobic parameters amino-acid side chains from partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem.-Chim. Ther. 18 369–375.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
