

Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis
- PMID: 15501831
- PMCID: PMC1360176
- DOI: 10.1074/mcp.M400094-MCP200
Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis
Abstract
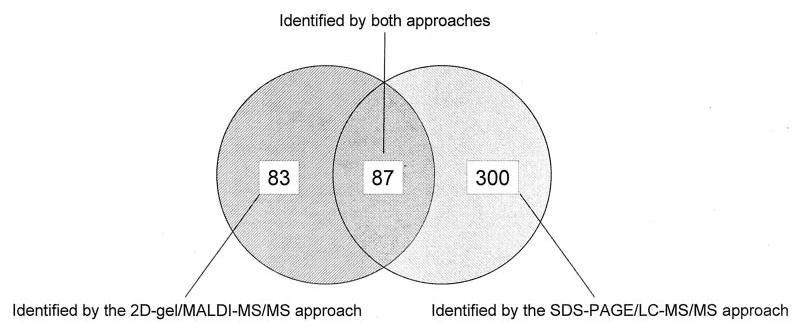
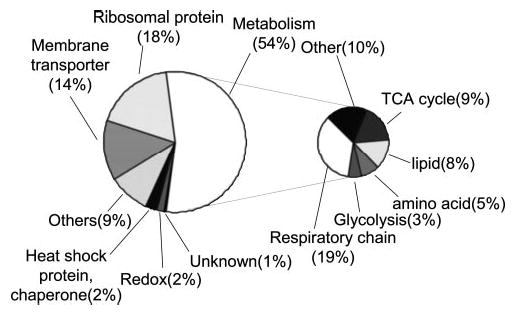
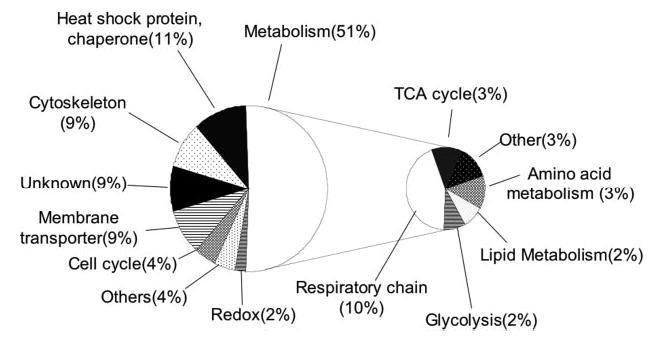
Mutations in copper-zinc superoxide dismutase (SOD1) have been linked to a subset of familial amytrophic lateral sclerosis (fALS), a fatal neurodegenerative disease characterized by progressive motor neuron death. An increasing amount of evidence supports that mitochondrial dysfunction and apoptosis activation play a critical role in the fALS etiology, but little is known about the mechanisms by which SOD1 mutants cause the mitochondrial dysfunction and apoptosis. In this study, we use proteomic approaches to identify the mitochondrial proteins that are altered in the presence of a fALS-causing mutant G93A-SOD1. A comprehensive characterization of mitochondrial proteins from NSC34 cells, a motor neuron-like cell line, was achieved by two independent proteomic approaches. Four hundred seventy unique proteins were identified in the mitochondrial fraction collectively, 75 of which are newly discovered proteins that previously had only been reported at the cDNA level. Two-dimensional gel electrophoresis was subsequently used to analyze the differences between the mitochondrial proteomes of NSC34 cells expressing wild-type and G93A-SOD1. Nine and 36 protein spots displayed elevated and suppressed abundance respectively in G93A-SOD1-expressing cells. The 45 spots were identified by MS, and they include proteins involved in mitochondrial membrane transport, apoptosis, the respiratory chain, and molecular chaperones. In particular, alterations in the post-translational modifications of voltage-dependent anion channel 2 (VDAC2) were found, and its relevance to regulating mitochondrial membrane permeability and activation of apoptotic pathways is discussed. The potential role of other proteins in the mutant SOD1-mediated fALS is also discussed. This study has produced a short list of mitochondrial proteins that may hold the key to the mechanisms by which SOD1 mutants cause mitochondrial dysfunction and neuronal death. It has laid the foundation for further detailed functional studies to elucidate the role of particular mitochondrial proteins, such as VDAC2, in the pathogenesis of familial ALS.
Figures



References
-
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. - PubMed
-
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. - PubMed
-
- Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. - PubMed
-
- Brown RH., Jr SOD1 aggregates in ALS: Cause, correlate or consequence? Nat Med. 1998;4:1362–1364. - PubMed
-
- Cleveland DW, Liu J. Oxidation versus aggregation—How do SOD1 mutants cause ALS? Nat Med. 2000;6:1320–1321. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
