

Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor
- PMID: 15507629
- PMCID: PMC525057
- DOI: 10.1128/JVI.78.22.12428-12437.2004
Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor
Abstract
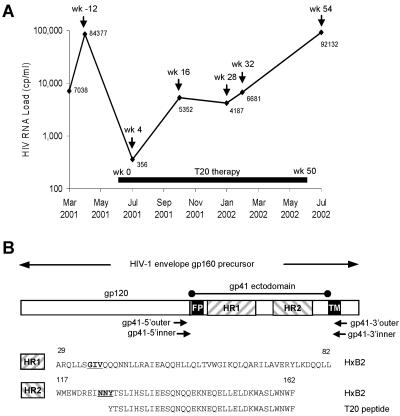
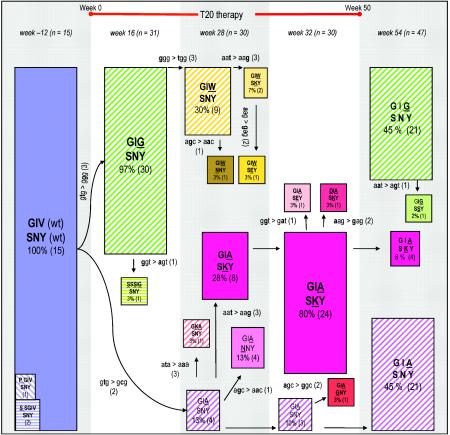
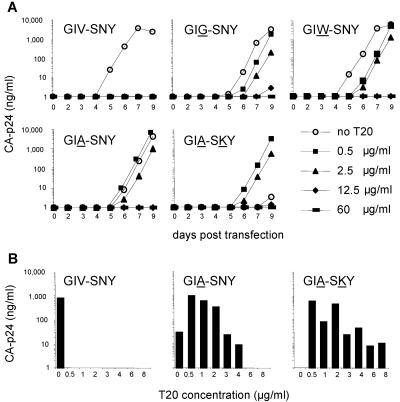
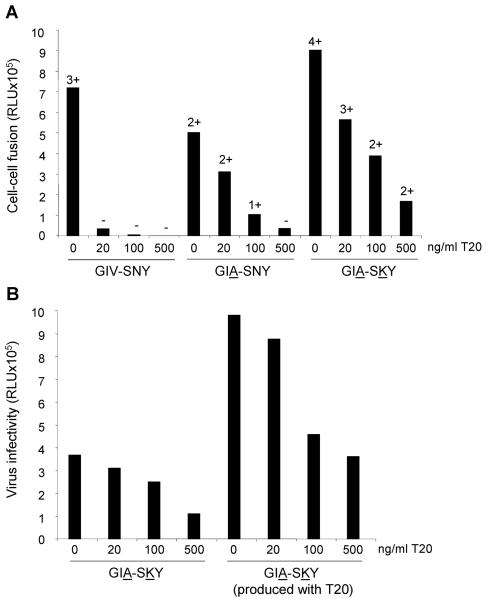
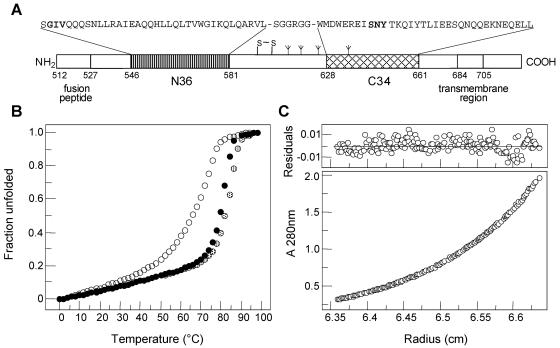
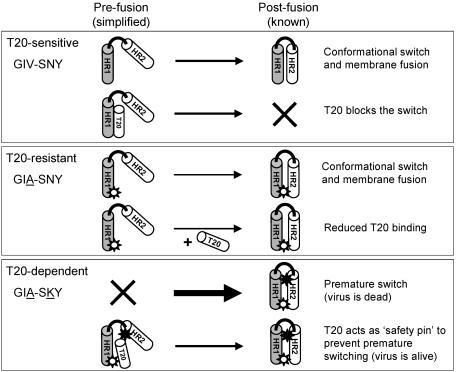
The fusion inhibitor T20 belongs to a new class of anti-human immunodeficiency virus type 1 (HIV-1) drugs designed to block entry of the virus into the host cell. However, the success of T20 has met with the inevitable emergence of drug-resistant HIV-1 variants. We describe an evolutionary pathway taken by HIV-1 to escape from the selective pressure of T20 in a treated patient. Besides the appearance of T20-resistant variants, we report for the first time the emergence of drug-dependent viruses with mutations in both the HR1 and HR2 domains of envelope glycoprotein 41. We propose a mechanistic model for the dependence of HIV-1 entry on the T20 peptide. The T20-dependent mutant is more prone to undergo the conformational switch that results in the formation of the fusogenic six-helix bundle structure in gp41. A premature switch will generate nonfunctional envelope glycoproteins (dead spikes) on the surface of the virion, and T20 prevents this abortive event by acting as a safety pin that preserves an earlier prefusion conformation.
Figures

References
-
- Baldwin, C. E., R. W. Sanders, and B. Berkhout. 2003. Inhibiting HIV-1 entry with fusion inhibitors. Curr. Med. Chem. 10:1633-1642. - PubMed
-
- Berkhout, B., A. T. Das, and N. Beerens. 2001. HIV-1 RNA editing, hypermutation and error-prone reverse transcription. Science 292:7. - PubMed
-
- Cantor, C., and P. Schimmel. 1980. Biophysical chemistry, part III. W. H. Freeman and Company, New York, N.Y.
-
- Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
