

A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system
- PMID: 15507682
- PMCID: PMC528815
- DOI: 10.1093/nar/gnh136
A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system
Abstract
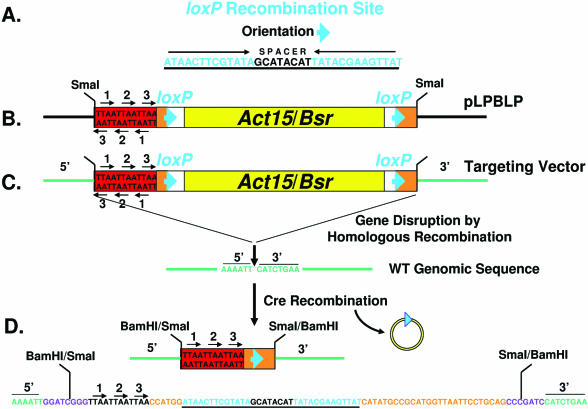
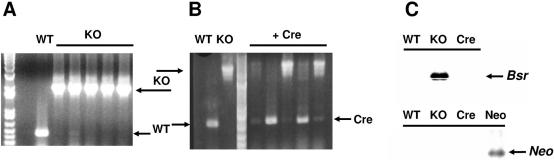
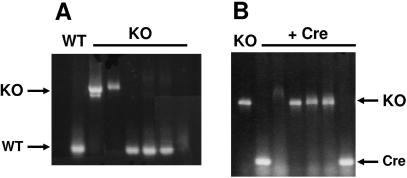
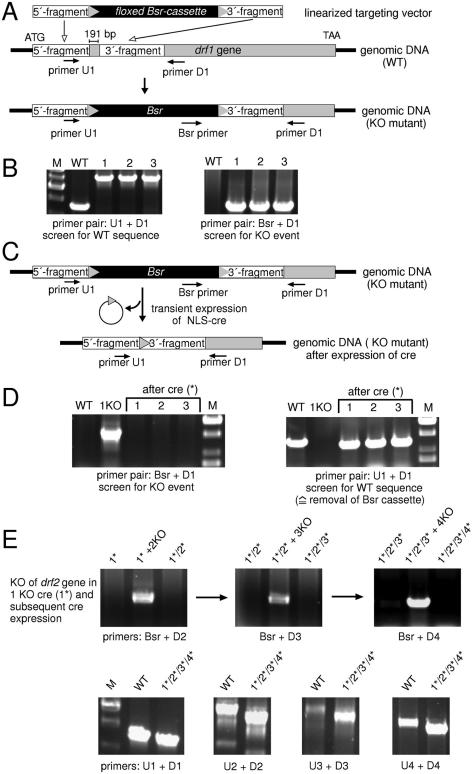
Dictyostelium discoideum has proven an exceptionally powerful system for studying numerous aspects of cellular and developmental functions. The relatively small ( approximately 34 Mb) chromosomal genome of Dictyostelium and high efficiency of targeted gene disruption have enabled researchers to characterize many specific gene functions. However, the number of selectable markers in Dictyostelium is restricted, as is the ability to perform effective genetic crosses between strains. Thus, it has been difficult to create multiple mutations within an individual cell to study epistatic relationships among genes or potential redundancies between various pathways. We now describe a robust system for the production of multiple gene mutations in Dictyostelium by recycling a single selectable marker, Blasticidin S resistance, using the Cre-loxP system. We confirm the effectiveness of the system by generating a single cell carrying four separate gene disruptions. Furthermore, the cells remain sensitive to transformation for additional targeted or random mutagenesis requiring Blasticidin selection and for functional expression studies of mutated or tagged proteins using other selectable markers.
Figures
Similar articles
-
Generation of multiple knockout mutants using the Cre-loxP system.Methods Mol Biol. 2006;346:187-99. doi: 10.1385/1-59745-144-4:187. Methods Mol Biol. 2006. PMID: 16957291
-
Highly effective removal of floxed Blasticidin S resistance cassettes from Dictyostelium discoideum mutants by extrachromosomal expression of Cre.Eur J Cell Biol. 2012 Feb;91(2):156-60. doi: 10.1016/j.ejcb.2011.11.001. Epub 2011 Dec 10. Eur J Cell Biol. 2012. PMID: 22154549
-
Blasticidin resistance cassette in symmetrical polylinkers for insertional inactivation of genes in Dictyostelium.Folia Biol (Praha). 1998;44(5):185-8. Folia Biol (Praha). 1998. PMID: 10732710
-
Cre/loxP recombination system and gene targeting.Methods Mol Biol. 2002;180:175-204. doi: 10.1385/1-59259-178-7:175. Methods Mol Biol. 2002. PMID: 11873650 Review. No abstract available.
-
The LoxP/CRE system and genome modification.Methods Mol Biol. 2001;158:83-94. doi: 10.1385/1-59259-220-1:83. Methods Mol Biol. 2001. PMID: 11236673 Review. No abstract available.
Cited by
-
Analysis of the Microprocessor in Dictyostelium: The Role of RbdB, a dsRNA Binding Protein.PLoS Genet. 2016 Jun 6;12(6):e1006057. doi: 10.1371/journal.pgen.1006057. eCollection 2016 Jun. PLoS Genet. 2016. PMID: 27272207 Free PMC article.
-
Identification of distinct biological functions for four 3'-5' RNA polymerases.Nucleic Acids Res. 2016 Sep 30;44(17):8395-406. doi: 10.1093/nar/gkw681. Epub 2016 Aug 2. Nucleic Acids Res. 2016. PMID: 27484477 Free PMC article.
-
Phospholipids containing ether-bound hydrocarbon-chains are essential for efficient phagocytosis and neutral lipids of the ester-type perturb development in Dictyostelium.Biol Open. 2020 Jul 16;9(7):bio052126. doi: 10.1242/bio.052126. Biol Open. 2020. PMID: 32675052 Free PMC article.
-
CP250, a novel acidic coiled-coil protein of the Dictyostelium centrosome, affects growth, chemotaxis, and the nuclear envelope.Mol Biol Cell. 2009 Oct;20(20):4348-61. doi: 10.1091/mbc.e09-03-0180. Epub 2009 Aug 19. Mol Biol Cell. 2009. PMID: 19692569 Free PMC article.
-
Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: a system for functional studies of the presenilin/gamma-secretase complex.Dis Model Mech. 2010 Sep-Oct;3(9-10):581-94. doi: 10.1242/dmm.004457. Epub 2010 Aug 10. Dis Model Mech. 2010. PMID: 20699477 Free PMC article.
References
-
- Aubry L. and Firtel,R. (1999) Integration of signaling networks that regulate Dictyostelium differentiation. Annu. Rev. Cell Dev. Biol., 15, 469–517. - PubMed
-
- Pang K.M., Lynes,M.A. and Knecht,D.A. (1999) Variables controlling the expression level of exogenous genes in Dictyostelium. Plasmid, 41, 187–197. - PubMed
-
- Barth C., Fraser,D.J. and Fisher,P.R. (1998) Co-insertional replication is responsible for tandem multimer formation during plasmid integration into the Dictyostelium genome. Plasmid, 39, 141–153. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
