

PITX2 gain-of-function in Rieger syndrome eye model
- PMID: 15509533
- PMCID: PMC1618668
- DOI: 10.1016/S0002-9440(10)63420-7
PITX2 gain-of-function in Rieger syndrome eye model
Abstract
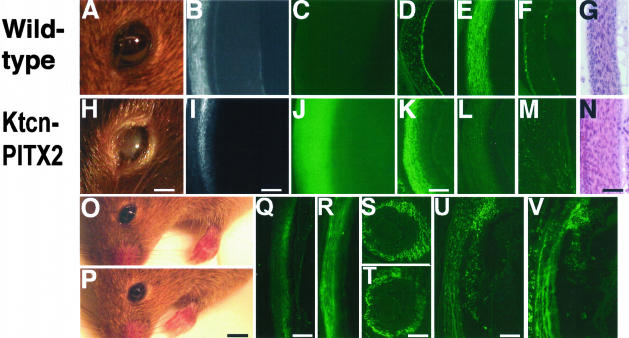
The human autosomal-dominant disorder Axenfeld-Rieger syndrome presents with defects in development of the eyes, teeth, and umbilicus. The eye manifests with iris ruptures, irido-corneal adhesions, cloudy corneas, and glaucoma. Transcription factors such as PITX2 and FOXC1 have been found to carry point mutations, causing the disorder. However, for approximately 40% of the cases, the pathogenesis is unknown. It has been reported that some mutations in PITX2 increase transactivation, whereas most mutations cause defects in DNA binding or transactivation. It is not known whether up-regulation of PITX2 activity can cause the disorder as well. Here we test this hypothesis directly by overexpressing PITX2A as a transgene in mouse corneal mesenchyme and iris, using keratocan-flanking sequences. The mice presented with corneal opacification, corneal hypertrophy, irido-corneal adhesions, and severely degenerated retina, resembling glaucoma. The corneal hypertrophy also resembles the corneal hypertrophy of Pitx2-/- mice. Control transgenic mice carrying point mutations T68P or K88E in PITX2A were normal. These findings indicate a novel pathogenetic mechanism in which excess corneal and iridal PITX2A cause glaucoma and anterior defects that closely resemble Axenfeld-Rieger syndrome.
Figures





References
-
- Clark AF, Yorio T. Ophthalmic drug discovery. Nat Rev Drug Disc. 2003;2:448–459. - PubMed
-
- Gage PJ, Suh H, Camper SA. The bicoid-related Pitx gene family in development. Mamm Genome. 1999;10:197–200. - PubMed
-
- Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002;11:1177–1184. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
